ARCIMBOLDO_SHREDDER Spherical tutorial
Aims of the tutorial
This tutorial shows how to use the spherical mode in ARCIMBOLDO-SHREDDER1. This mode generates, starting from a distant homolog template that is annotated in terms of secondary and tertiary structure, a set of compact search models used as a library in ARCIMBOLDO BORGES, profiting from specific options in PHASER and SHELXE for model improvement. This example uses the latest ARCIMBOLDO_SHREDDER as of July 2018.
Experimental details and data
Test data for our tutorial have been obtained from the protein Slt (PDB ID: 5OHU). It is a lytic transglycosylase from Pseudomonas aeruginosa. This structure was solved2 with data extending to 2.1 Å. The asymmetric unit contains one monomer of 642 residues.
Data are summarized in the following table:
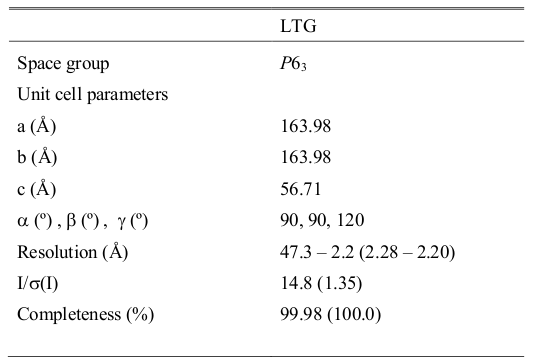
Figure 1. Slt Structure colored after B-value.
Step by Step tutorial
1. Selection of a starting template
A BLAST3 search of the LTG sequence against the PDB finds a model, 1QSA, with 33% of sequence identity over a range of 603 amino acids.
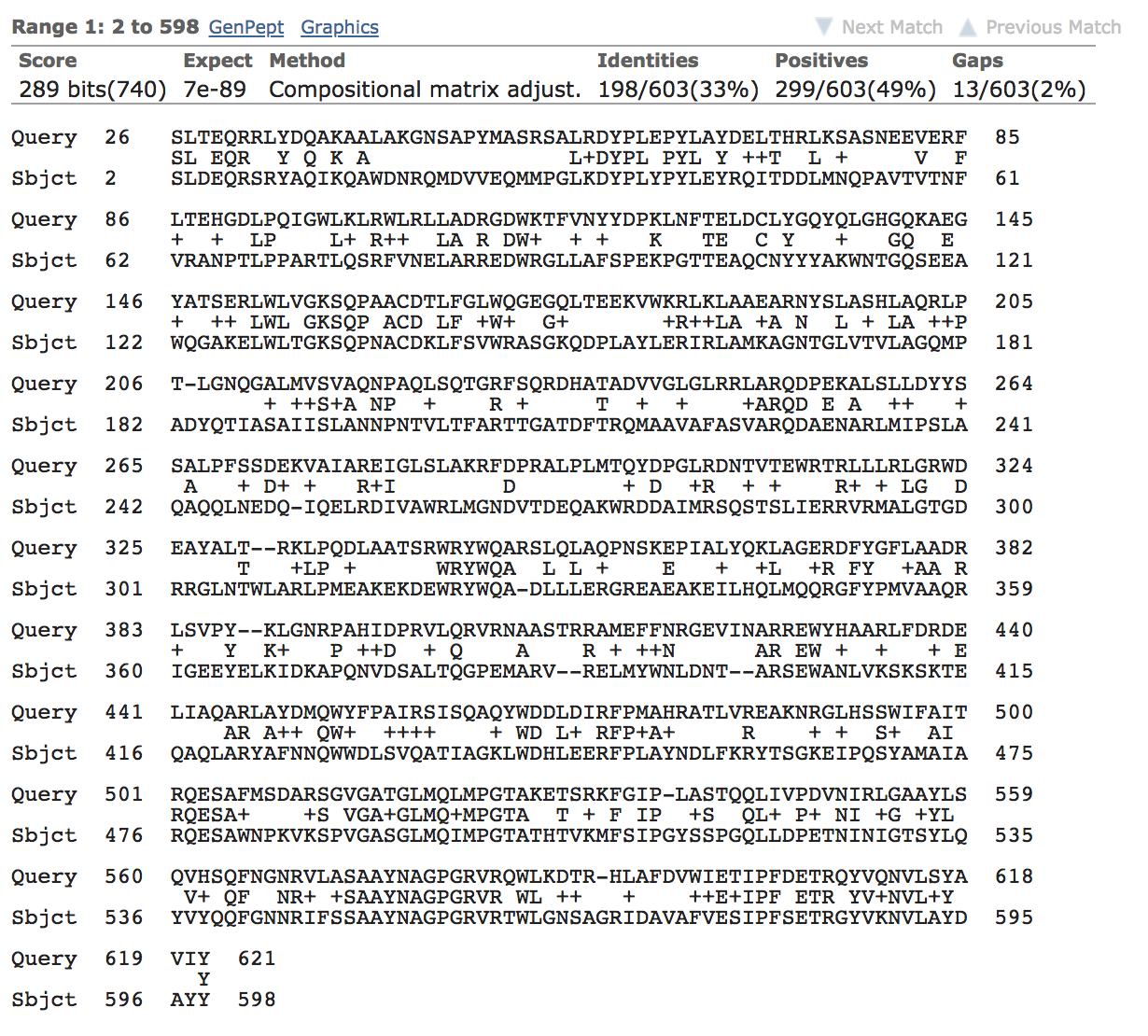
Figure 2. LTG alignment against 1QSA
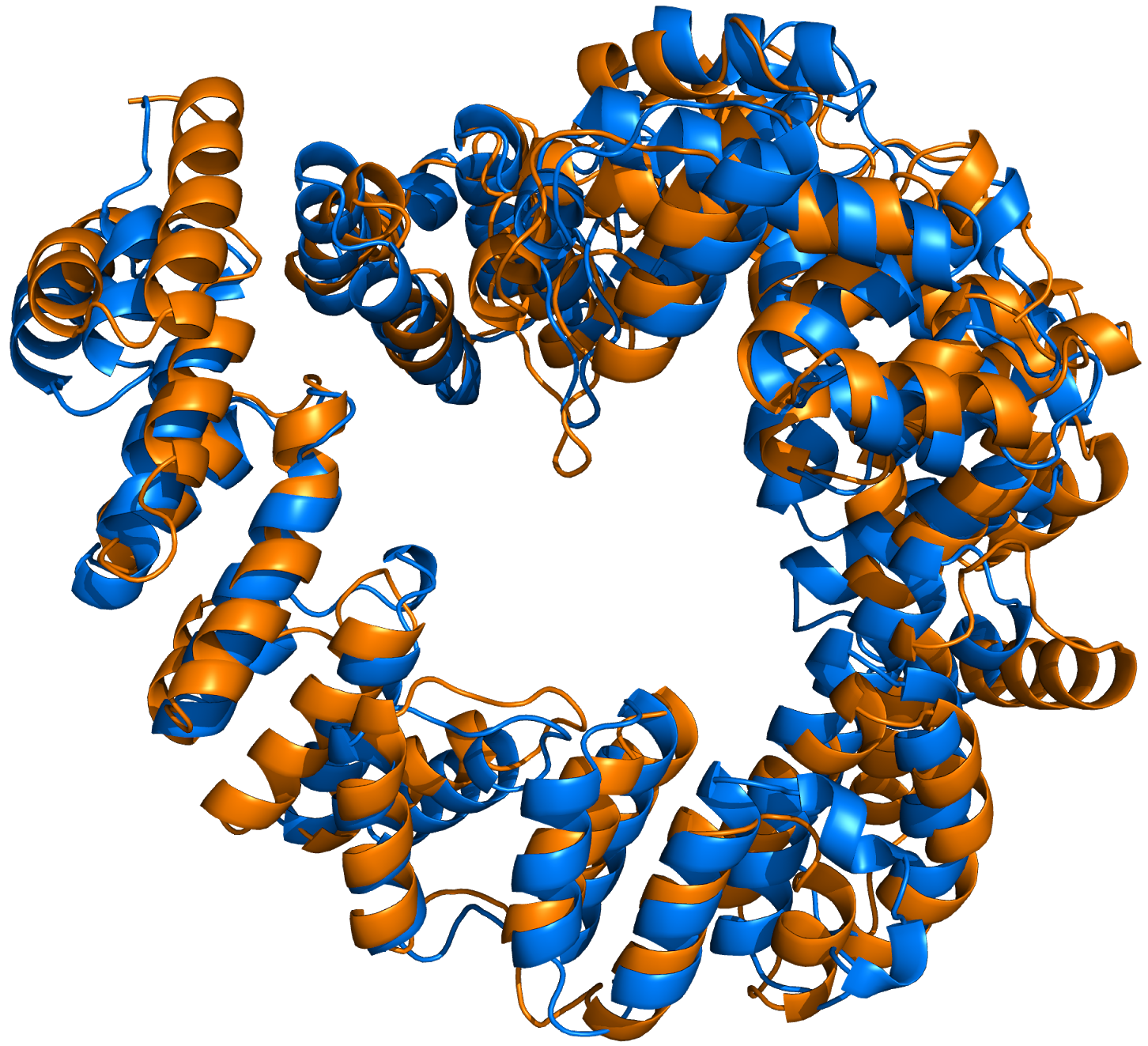
Figure 3. LTG final structure (blue) superposed with 1QSA template (orange).
A classical molecular replacement approach with the 1QSA model fails due to the high deviation between the model and the final structure, which presents an overall similar core but many local differences. In fact, the r.m.s.d. between both structures is 4.6 Å over a core of 582 Ca atoms (Figure 3)
Required Input
All required files can be downloaded here.
The description of the configuration file follows:
[CONNECTION]: distribute_computing: local_grid setup_bor_path: /path/to/setup.bor [LOCAL] path_local_phaser: /path/to/local_phaser path_local_shelxe: /path/to/local_shelxe [GENERAL]: working_directory: /path/to/working_directory mtz_path: %(working_directory)s/5ohu.mtz hkl_path: %(working_directory)s/5ohu.hkl [ARCIMBOLDO-SHREDDER] name_job: 5ohu_spheres molecular_weight: 70153 f_label: F sigf_label: SIGF number_of_component: 1 model_file: /absolute/path/to/1qsacut.pdb SHRED_METHOD: spherical rmsd_shredder: 1.2 alixe: True
In the .bor file you need to specify:
In the [ARCIMBOLDO-SHREDDER] section you will need to define the contents of the asymmetric unit, the model pdb to use, and the labels for the mtz file. Other parameters that have defaults but that maybe changed are the expected rmsd of the models (rmsd_shredder), the definition of their size (sphere_definition), and the settings of model refinement strategies such as gyre and gimble. In this case, we will use default values. The starting rmsd plays a very relevant role in shredder, as it is used both for selecting the size of the models since it conditions the eLLG4 and PHASER’s target functions in the searches. Moreover, in successive cycles of gyre and gimble refinement the rmsd is decreased by 0.2. Thus, the rmsd used in translation search and rigid body refinement will typically be the set rmsd -0.2 Å. Therefore, it is a parameter worth exploring in case the defaults render no solution. Also it is worth mentioning that it refers to the expected rmsd for the models, not the full template. Starting rmsd values within the range of 1.2 to 0.8 might be appropriate. A recommended optimisation is the activation of the phase combination strategy implemented in ALIXE5. If set to True, a step of combination of partial solutions in reciprocal space will be performed. A complete description of all optional and mandatory parameters can be found in the manual.
Execution
You can run the program from the ccp4i interface or from a terminal, redirecting the output to a log file. This tutorial describes use through a command file, as interfaces are self-explanatory.
1. Interactively
ARCIMBOLDO_SHREDDER 5ohu.bor
2. In background
nohup ARCIMBOLDO_SHREDDER 5ohu.bor >& log &
Output and Results
In the directory where you launched ARCIMBOLDO_SHREDDER, you will find a directory called models containing a library of pdbs. Around each Calpha in the template, models are cut in a spatial way, producing a set of non-redundant, overlapping, compact models. In the default mode, they are also annotated in different chains in order to decompose them and perform gyre and gimble6 refinement, aiming to give more degrees of freedom and obtain a more accurate model.
The directory called ARCIMBOLDO_BORGES contains the output of the ARCIMBOLDO_BORGES run using the library in models.The html output of the library search is found here. The first section echoes all parameters used for the run, so that defaults are listed along with values for the parameters set through the .bor file. This allows to reproduce the run even if defaults may change in future versions.The next section displays a graph and a table summarizing the rotation clustering step as in ARCIMBOLDO_BORGES7. A sortable table follows, summarizing the results for all PHASER and SHELXE steps, including top and average figures of merit for each rotation cluster that has been evaluated.
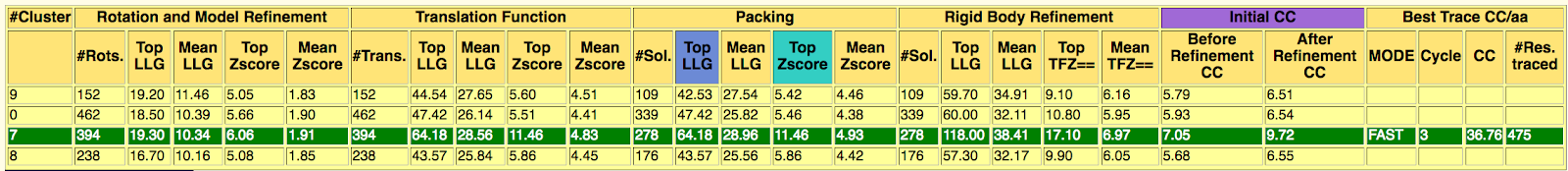
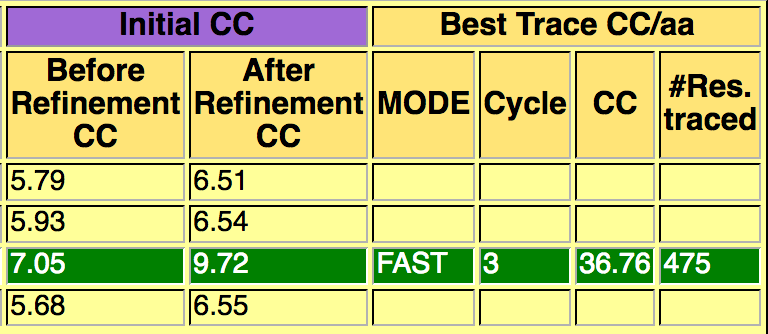
The last section of the html contains links to access the best current solution: the pdb of the traced structure and the map in .phs format. In this case, a map coming from a combination of 61 solutions, using as a reference the solution produced in rotation cluster 7 by the model obtained using as center the residue 589, produces a complete interpretable map after density modification and autotracing with SHELXE8. Figure 4 shows a detail of the map before density modification (orange map) and autotracing and after (blue map). A correlation coefficient of 36.76% with 475 residues traced is obtained.
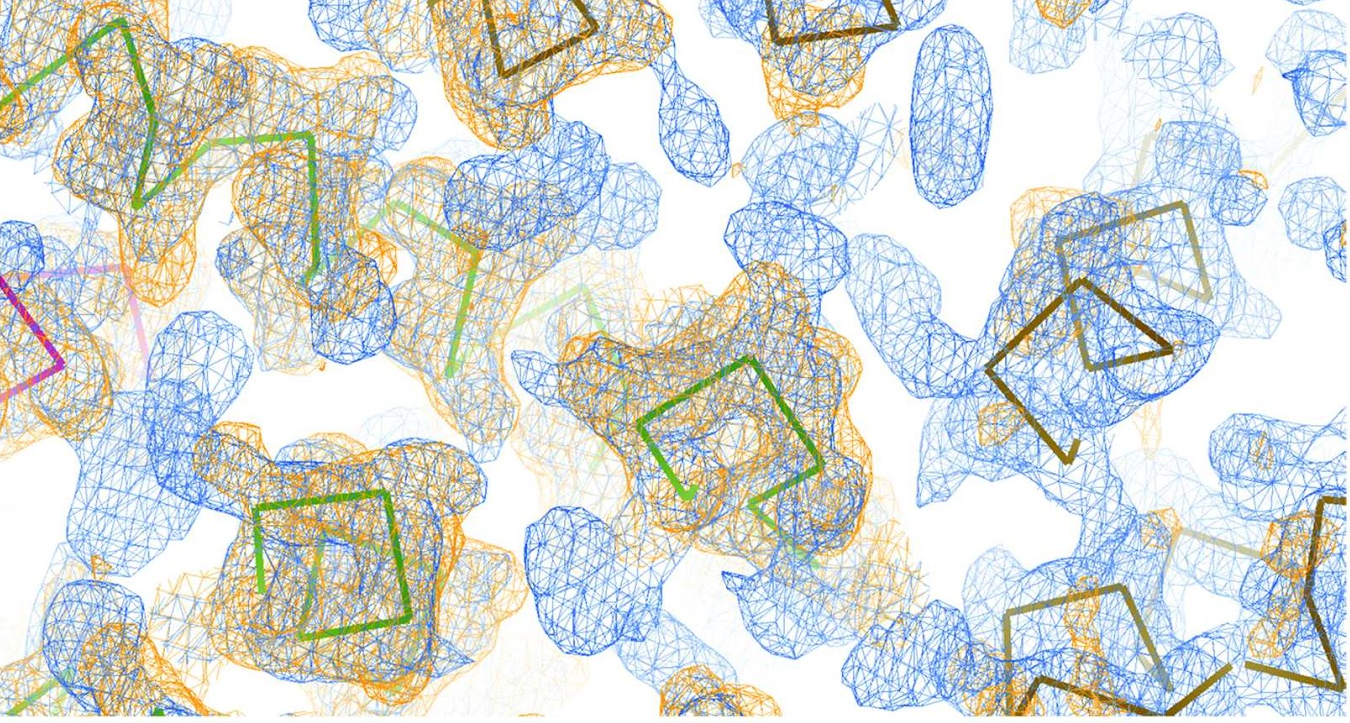
Figure 4. A detail of the electron density map before autotracing (orange) and after (blue).
References
-
Exploiting distant homologues for phasing through the generation of compact fragments, local fold refinement and partial solution combination.
Millán, C. , Sammito, M. D., McCoy, A. J., Nascimento, A. F., Petrillo, G., Oeffner, R. D., Domínguez-Gil, T. , Hermoso, J. A., Read, R. J. and Usón, I.
Acta Cryst. D74: 290-304 (2018) (doi:10.1107/S2059798318001365)
-
Exolytic and endolytic turnover of peptidoglycan by lytic transglycosylase Slt of Pseudomonas aeruginosa.
Mijoon Lee, María T. Batuecas, Shusuke Tomoshige, Teresa Domínguez-Gil, Kiran V. Mahasenan, David A. Dik, Dusan Hesek, Claudia Millán, Isabel Usón, Elena Lastochkin, Juan A. Hermoso and Shahriar Mobashery.
Proceedings of the National Academy of Sciences Apr 2018, 115 (17) 4393-4398 (doi:10.1073/pnas.1801298115)
-
Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.
Altschul SF1, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W and Lipman DJ.
Nucleic Acids Res. 1997 Sep 1;25(17):3389-402 (doi:10.1093/nar/25.17.3389)
-
Ab initio solution of macromolecular crystal structures without direct methods.
Airlie J. McCoy, Robert D. Oeffner, Antoni G. Wrobel, Juha R. M. Ojala, Karl Tryggvason, Bernhard Lohkamp and Randy J. Read.
PNAS April 4, 2017. 114 (14) 3637-3641 (doi:10.1073/pnas.1701640114)
-
Combining phase information in reciprocal space for molecular replacement with partial models.
Millan, C., Sammito, M., Garcia-Ferrer, I., Goulas, T., Sheldrick, G. M. and Uson, I.
Acta Cryst. D71, 1931-1945 (doi:10.1107/S1399004715013127)
-
Gyre and gimble: a maximum-likelihood replacement for Patterson correlation refinement.
McCoy, A. J., Oeffner, R. D., Millan, C., Sammito, M., Uson, I. and Read, R. J.
Acta Cryst. D74, 279-289 (doi:10.1107/S2059798318001353)
-
Exploiting tertiary structure through local folds for crystallographic phasing.
Massimo Sammito, Claudia Millán, Dayté D Rodríguez, Iñaki M de Ilarduya, Kathrin Meindl, Ivan De Marino, Giovanna Petrillo, Rubén M Buey, José M de Pereda, Kornelius Zeth, George M Sheldrick and Isabel Usón.
Nature Methods volume 10, pages 1099–1101 (2013) (doi:10.1038/nmeth.2644)
-
An introduction to experimental phasing of macromolecules illustrated by SHELX; new autotracing features.
Uson, I. and Sheldrick, G. M.
Acta Cryst. D74, 106-116. (doi:10.1107/S2059798317015121)