DNA Tutorial for Arcimboldo
Aims of the tutorial
This tutorial shows how to launch BORGES-ARCIMBOLDO in the particular case of DNA-binding protein libraries (Pröpper et al., 2014). In the present case a Zinc-finger library is used to phase the structure of the Krueppel-like factor 4 (Klf4) (Schuetz et al., 2011) that contains the Zinc-finger domain.
This example uses the latest ARCIMBOLDO release, but the previous tutorial is accesible here
Step summary
1) Create a library of model templates
2) Use the library against the diffraction data to solve the structure with ARCIMBOLDO_BORGES
Experimental details
Klf4 is a zinc-finger transcription factor indispensable for terminal maturation of epithelial tissues, and it is also involved in the generation of pluripotent embryonic stem cells from differentiated tissues. This structure is deposited in the Protein Data Bank under the PDB code 2WBS.
Details of the data are summarized in the following table:
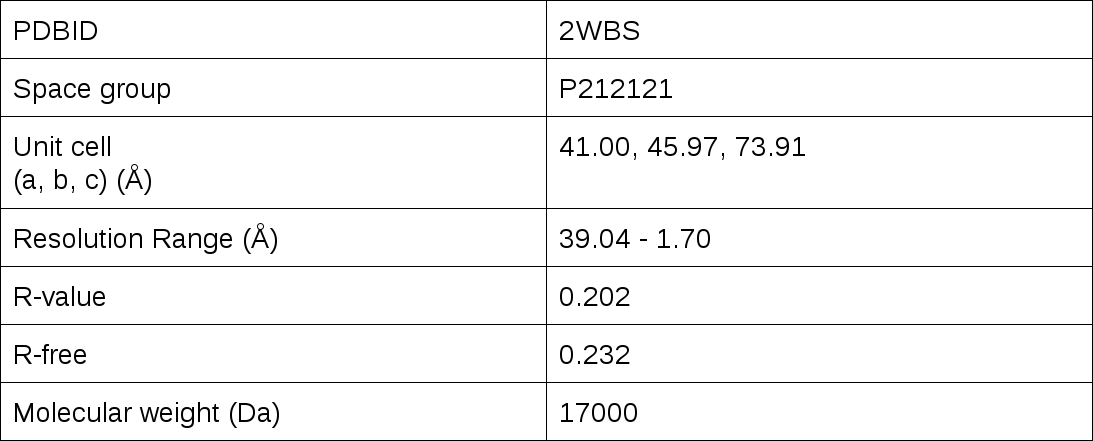
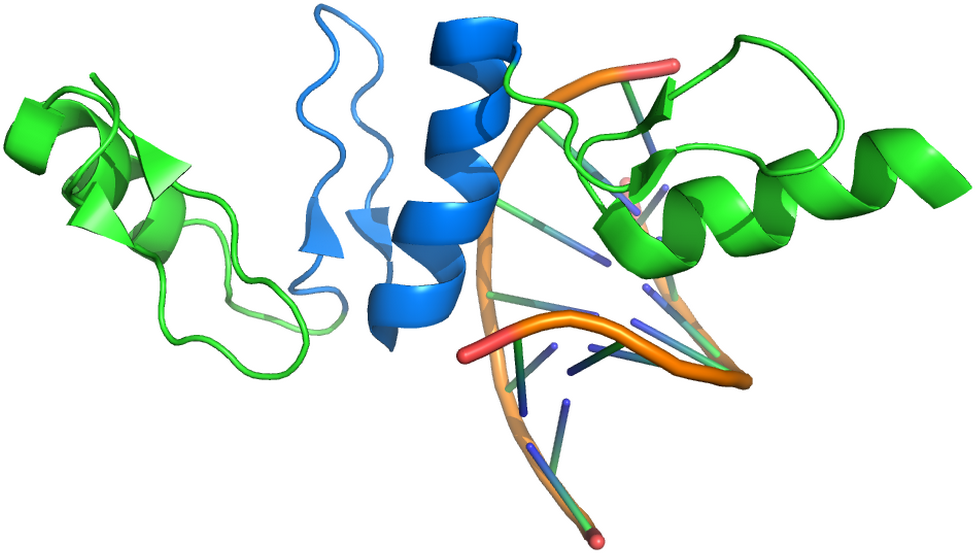
Figure 1. Cartoon representation of the Klf4 structure binding to the DNA.The Zinc-finger motif (shown in marine blue) appears three times in the structure. The motif is characterized by a short helix and two short beta-stranded segments connected by a loop.
A classical ARCIMBOLDO approach using a single helix as search model is not indicated because of the short length of the helix in the motif. Instead, exploiting a library of superposed models could take into account the local variability and improve the chance of phasing the structure.
Step by Step tutorial
1. Creation of the library from a selected search model
To create a library in BORGES a pdb model is needed as input for the program to define the profile of the geometry of the fold, which is required to extract it from the whole database. The user can define critical thresholds such as angles and distances between fragments and configure a certain degree of deviation from the input search model. In this case we want to extract a library for Zinc-finger motifs. Here, the fold from the PDB entry 1UBD is chosen as a search model (Houbaviy et al., 1996). This entry corresponds to the human YY1 Zinc-finger domain bound to the Adeno-associated virus P5 initiator element.
Figure 2. Cartoon representation of the 1UBD deposited structure that contains the human YY1 Zinc-finger domain bound to the DNA. a) in red is highlighted the portion used as search model to create the library with BORGES; b) rainbow color representation of the B-factors of the deposited protein data, colors ranging from red to blue with red representing high values and blue low values of the B-factors.
As shown in Figure 2, the selected region (2.a) is not necessarily the most rigid one (2.b), which in general is easier to locate. Moreover, the RMSD of the search model vs. the final structure is higher than 0.5 Å, which is what is usually required to phase from such a small model. This does not represent a big problem because it is possible to extract a library of similar folds that could contain models close enough to the target, and from which the correct phases can be bootstrapped. The model is 27 aa in length, and it is characterized by a short helix of 14 aa, two very short antiparallel strands of variable length and a specific loop region that is also conserved in the fold.
Due to the role of the loop in the fold, the library is created with a novel algorithm in BORGES that allows to extract not only alpha helices and beta strands, but also coils and loops.
This new algorithm is still under development and will be described soon, so for this case the created library can be downloaded here.
Output
The BORGES library that is extracted has the following characteristics:
- Models are superimposed against the template used for creation of the library
- B-factors are set to the same value (25.00)
- Atoms with an occupancy under 50% are omitted
- PDB residues are renumbered when extracting fragments from the original PDB in the database, but a remark is added so that original residues are tracked
- Residue numbers for separated secondary structure elements are non-contiguous
In this example, the library was created against a subset of Zinc-finger proteins and the resulting set was not clustered. The library contains 45 models.
2. Usage of the library against experimental data to solve the structure
Data preparation and data conversion
Experimental data in both mtz and hkl formats have to be provided. If you have an hkl file you can use the programs F2MTZ and TRUNCATE or generate a .sca file and use SCALEPACK2MTZ to create an mtz file. On the contrary, if you have an .mtz file you can use MTZ2HKL to get your .hkl file.
Input
DNA input files and DNA library files- An .mtz file containing the reflection data
- A SHELX reflection file .hkl containing the reflection data
- The configuration .bor file
- A library created with BORGES
Here it follows the description of the configuration file:
[CONNECTION]: distribute_computing: local_grid setup_bor_path: /path/to/setup.bor [GENERAL]: working_directory: /path/to/working_directory mtz_path: %(working_directory)s/2wbs.mtz hkl_path: %(working_directory)s/2wbs.hkl [BORGES-ARCIMBOLDO] name_job: 2wbs molecular_weight: 17000 number_of_component: 1 f_label: F sigf_label: SIGF shelxe_line: -m30 -v0 -y1.9 -a50 -t20 -e1.0 -q -s0.45 clusters: all library_path: ./zfing_lib
The GENERAL section contains information about the location of the required and provided I/O paths.
As shown in the example file it is possible to create variables inside the .bor file that will be visible only from other tags in the same section. They could be used to avoid repeating long paths multiple times. In the example a variable working_directory is created by using the assignment (=) symbol after its nameworking_directory= /path/to/the/working/directory/
Then it is used for the following tags by enclosing the variable name by the symbols %(name)smtz_path: %(working_directory)s/2wbs.mtzhkl_path: %(working_directory)s/2wbs.hkl
To use ARCIMBOLDO with a BORGES library it is necessary to create the section ARCIMBOLDO-BORGES, in which it is required to insert the name of the job that will be also be the name of the created html file, and information related to the protein structure itself. Molecular weight and number of components in the asymmetric unit will be provided to PHASER for computing correct statistics against the Maximum Likelihood function. The user can parameterize the estimation of the errors for the Log-Likelihood function by increasing the default Root Mean Square Deviation by using the rmsd tag in this section. For more details see the ARCIMBOLDO tutorial or the manual.
It is also required to provide the correct label identification for the amplitudes (F) and their sigmas (SIGF) used in the provided mtz file. In fact this file supports multiple data sets and it is necessary to explicitly choose what set you would like to use for the case. An mtz file is a binary file but programs like MTZDMP available with the CCP4 suite can display their content.
The user can also specify the parametrization line used for the program SHELXE at the density modification and autotracing step of the procedure. If it is not specified BORGES will configure a default one. In this particular case it was fundamental to increase both the parameter -m (number of density modification cycles) and the parameter -t (time factor for searching tripeptides) to phase the structure. Be aware that even if the parameter -a (number of autotracing cycles) is set to 50, BORGES will perform each autotracing cycle as an independent step and it will automatically stop when it recognizes a solution.
Execution
To launch the BORGES-ARCIMBOLDO you can again choose between:
1. Interactively:
ARCIMBOLDO_BORGES conf_file.bor
2. In background:
ARCIMBOLDO_BORGES conf_file.bor < password_file >& log
Output: Did it work? What may I change?
In the directory where you launched BORGES, an html file called like the job is created (2wbs.html). It is written while the program is running and updated as results are obtained. You may use this information for manual intervention (stopping the run, reparameterization). If you leave it running, BORGES will sequentially try the best clusters (printed in green in the html, based on the figures of merit). Once a solution is found (SHELXE traced mainchain CC > 25%), it stops after some recycling steps to improve it.
Analyzing the Output
The html output is the file that summarizes your job instructions and results. At the beginning the full configuration file will be reported (Fig. 3) showing not only the parametrization that the user configured but also all the defaults that remained unchanged.
A detailed explanation of the keywords can be found in the manual.
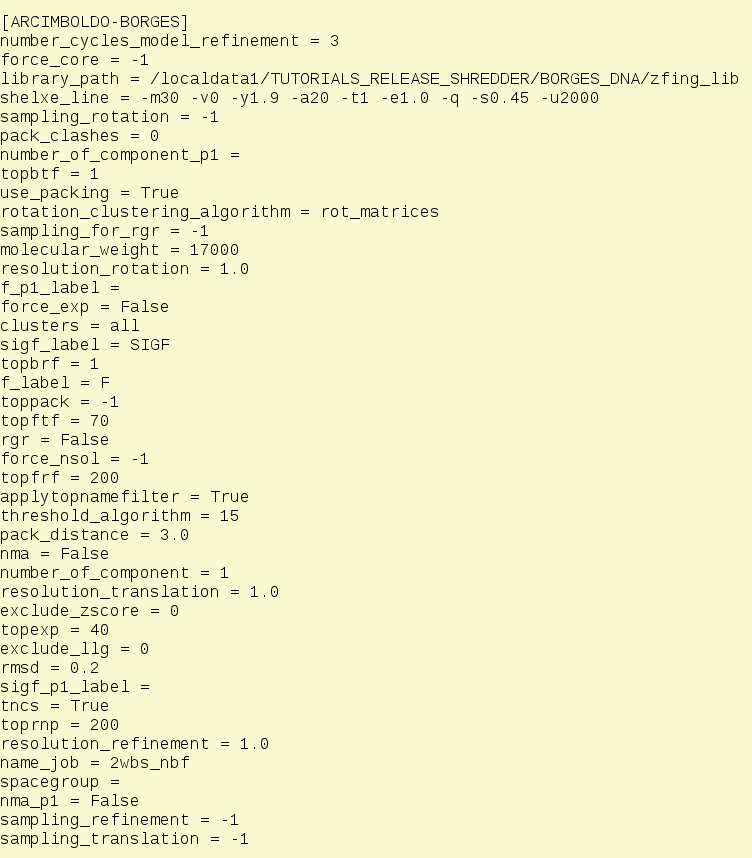
Figure 3. View of all the full keywords used for the run. This information can be used to rerun the job even if the internal defaults of the program are changed during the version releases.
The file follows with a small summary of the data (Fig. 4) where the information about the space group, resolution and number of reflections are shown. In this section BORGES will display also special warnings when unusual configuration is used or when resolution range of the data could be problematic for the effectiveness of the method. Following this summary there is a table in which the rotation clusters found are listed. By clicking on “Hide not relevant” the full list of clusters will appear, but BORGES automatically selects the most promising ones that are going to be used for further steps.
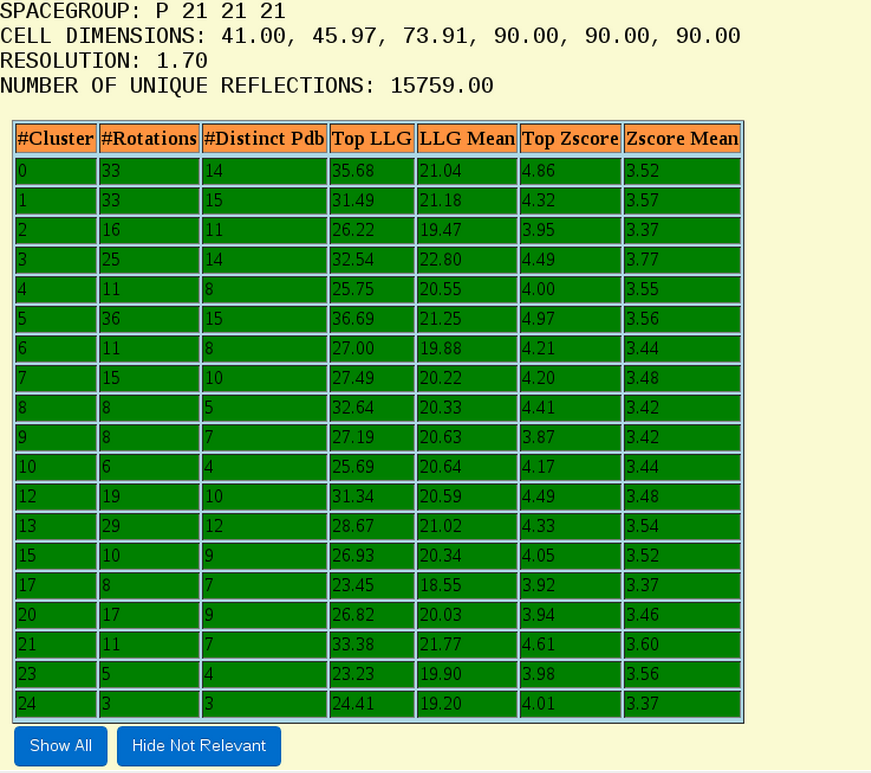
Figure 4. View of the summary of the data and the rotation clusters table. In this view only the “green” clusters are shown. Note that all rows of all tables can be sorted numerically by columns by just clicking at the column label.
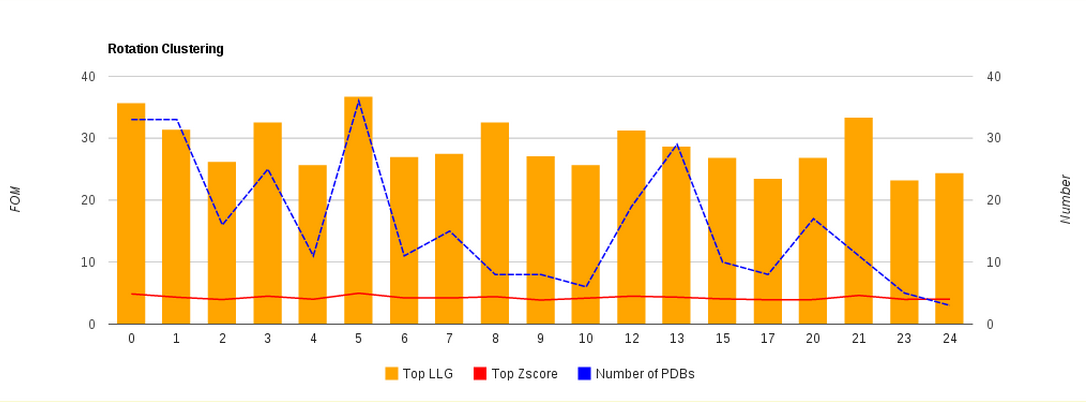
Figure 5. View of the cluster histograms.
Right to the previous table is printed a histogram graph in which each rotation cluster is shown with its highest LLG value, Z-score and the number of models (of the library) that it contains (Fig. 5).
Due to the amount of models and possible solutions BORGES analyzes all the candidate clusters and evaluates the figures of merit at run time. By evaluating statistics among all clusters the program proceeds to select a subset of clusters for being sent to the step of density modification and autotracing with SHELXE.
In Fig. 6 is shown a part of the table in which each row corresponds to a cluster and relative FOMs are reported for each step. In this example the high Z-score in the packing step and the highest SHELXE correlation coefficient indicates this cluster to be the first to which density modification and autotracing will be applied. The highlighting green line and the high CC indicate that BORGES has successfully phased the structure.
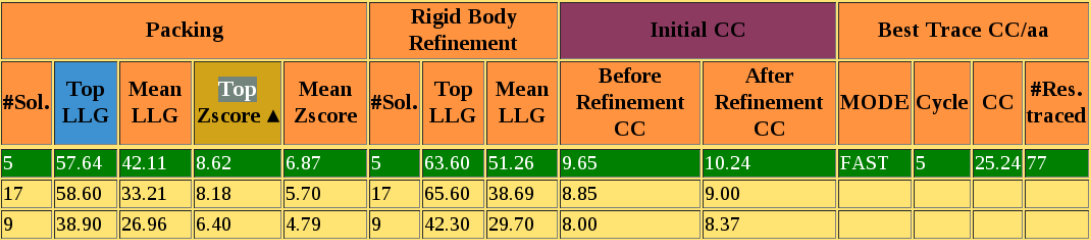
Figure 6. View of a part of the table highlighting the final CC obtained.
At the bottom of the page the user can find a backtracing of all the steps for the model in the library which leads to a correct solution (Fig.7).
All FOMs are reported and compared against the top of the cluster to which the solutions belongs.

Figure 7. Backtracing of the model that leads to a correct solution. When BORGES has solved the structure it also links to the final solution (in .pdb format) and the corresponding map (in .phs format) that can be used then for further refinement (DNA Output).
References
-
Cocrystal structure of YY1 bound to the adeno-associated virus P5 initiator.
Houbaviy, H. B., Usheva, A., Shenk, T. and Burley, S. K.
PNAS. 93, 13577-13582. (1996) (doi:10.1073/pnas.93.24.13577)
-
Structure solution of DNA-binding proteins and complexes with ARCIMBOLDO libraries.
Pröpper, K., Meindl, K., Sammito, M., Dittrich, B., Sheldrick, G. M., Pohl, E. and Usón, I.
Acta Cryst. D70, 1743-1757 (2014) (doi:10.1107/S1399004714007603)
-
The structure of the Klf4 DNA-binding domain links to self-renewal and macrophage differentiation.
Schuetz, A., Nana, D., Rose, C., Zocher, G., Milanovic, M., Koenigsmann, J., Blasig, R., Heinemann, U. and Carstanjen, D.
ACell. Mol. Life Sci. 68 , 3121-31 (2011) (doi:10.1007/s00018-010-0618-x)