SLIDER Secondary-Homolog tutorial
Aims of the tutorial: SEQUENCE SLIDER in Secondary-Homolog Mode
To illustrate SEQUENCE SLIDER in Secondary-Homolog Mode, the structure of Formaldehyde-Responsive Regulator (FrmR) E64H variant from Salmonella enterica serovar Typhimuriumwas chosen (Borges et al., 2020). It is a coiled coil with 91 residues and its data (PDB id: 5lcy) diffracts to 2.12 Å resolution in P21 space group with a solvent content of 42% and a tetramer in the asymmetric unit (Osman et al., 2016). It is possible to obtain its partial solution using ARCIMBOLDO_LITE in coiled coil mode (Caballero et al., 2018) using three polyalanine helices of 18 residues. Four partial solutions led upon expansion to a CC of above 40%. As for other structures with strong modulation of the data, such as coiled coils or nucleic acids, wrong solutions may be characterized by extremely high CC values, even above 40%, and successful side-chain discrimination may be used to disambiguate. Herein, top solution scored a CC of 45.3% with unconnected helices that summed 240 residues, corresponding to 64% of complete main chain structure.
PSIPRED predicts FrmR E64H sequence to have three helices composed of 26, 31 and 19 residues, separated by 4-6 coil residues. Such secondary structure prediction can be used to restrict SEQUENCE SLIDER sequence hypotheses into the helices traced with ARCIMBOLDO_LITE and find the correct sequence assignment.
# PSIPRED HFORMAT (PSIPRED V3.3) Conf: 989876688999888776569999999974798839999999996999878999999988 Pred: CCCCHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHHHHHHHHHHHHHH AA: MPHSPEDKKRILTRVRRIRGQVEALERALESGEPCLAILQQIAAVRGASNGLMSEMVEIH 10 20 30 40 50 60 Conf: 9653318999935888889999999998309 Pred: HHHHHCCCCCCHHHHHHHHHHHHHHHHHHHC AA: LKDHLVSGETTPDQRAVRMAEIGHLLRAYLK 70 80 90 Fragment 1: helix of length 26 5 PEDKKRILTRVRRIRGQVEALERALE 30 Fragment 2: helix of length 31 35 CLAILQQIAAVRGASNGLMSEMVEIHLKDHL 65 Fragment 3: helix of length 19 72 PDQRAVRMAEIGHLLRAYL 90
Step by Step tutorial
Input
We will need:
All required files can be downloaded here.
The configuration .bor file with the parameters for an SEQUENCE SLIDER run, which is defined as follows:
[CONNECTION]: distribute_computing: multiprocessing [GENERAL]: working_directory: /home/user/SLIDER-SSM/5lcy mtz_path: /home/user/SLIDER-SSM/5lcy/5lcy.mtz hkl_path: /home/user/SLIDER-SSM/5lcy/5lcy.hkl pdb_path: /home/user/SLIDER-SSM/5lcy/ARC_LITE_CC.pdb [SLIDER] secstr_path: /home/user/SLIDER-SSM/5lcy/5lcy.psipass2 molecular_weight: 10194 number_of_component: 4 f_label: F sigf_label: SIGF i_label: IMEAN sigi_label: SIGIMEAN rfree_label: FreeR_flag refinement_program = refmac sliding_tolerance: 4 chosen_chains: A;B;C;D RandomModels: 10 number_shelxe_trials: 0 [LOCAL] path_local_shelxe: /usr/local/ccp4-7.1/bin/shelxe path_local_refmac: /usr/local/ccp4-7.1/bin/refmac5 path_local_sprout: /usr/local/sprout_linux path_local_edstats: /usr/local/ccp4-7.1/bin/edstats
Execution
Interactively:
SLIDER.py 5lcy.bor
Output
In the HTML, the graphs show the statistics of sequence hypotheses after refinement by chosen chain (A, B, C and D) to have their helices side chain assigned:
- in the title, the chain followed by its respective residues whose side chain are being assigned.
- in the Y axis, sequence score, which describes the concordance of the sequence hypothesis in respect to the alignment.
- in X axis, log-likelihood gain (LLG) calculated by PHASER.
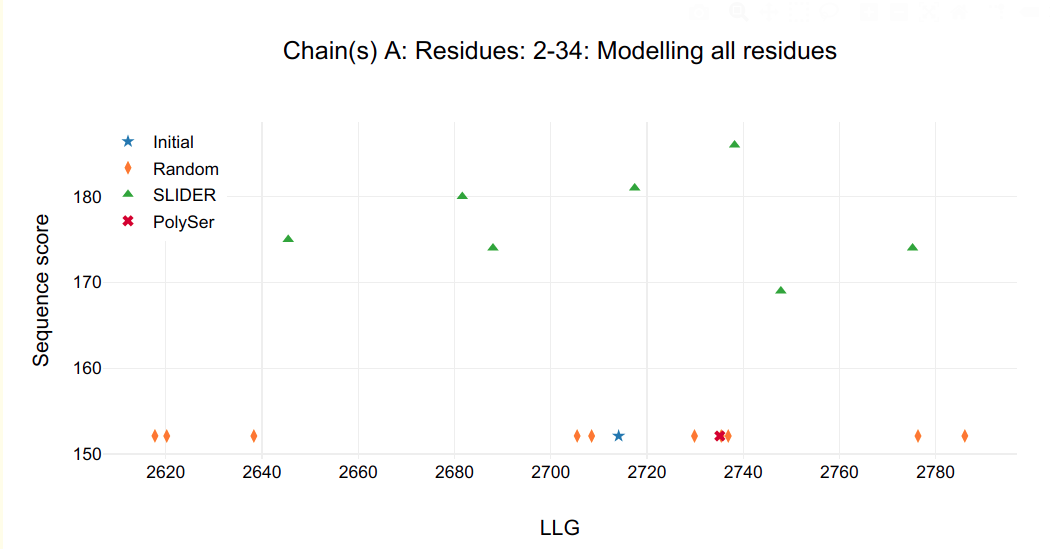
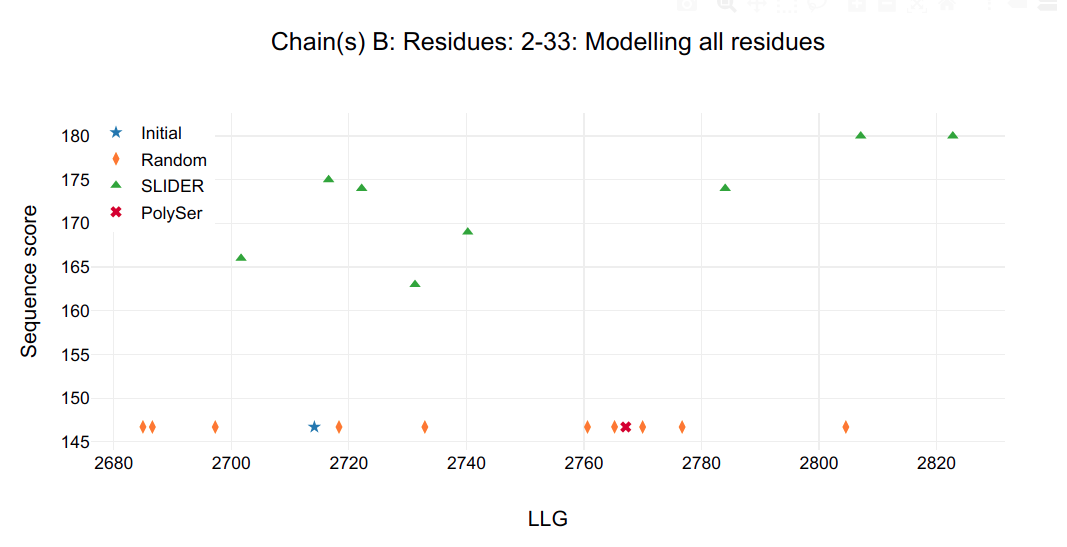
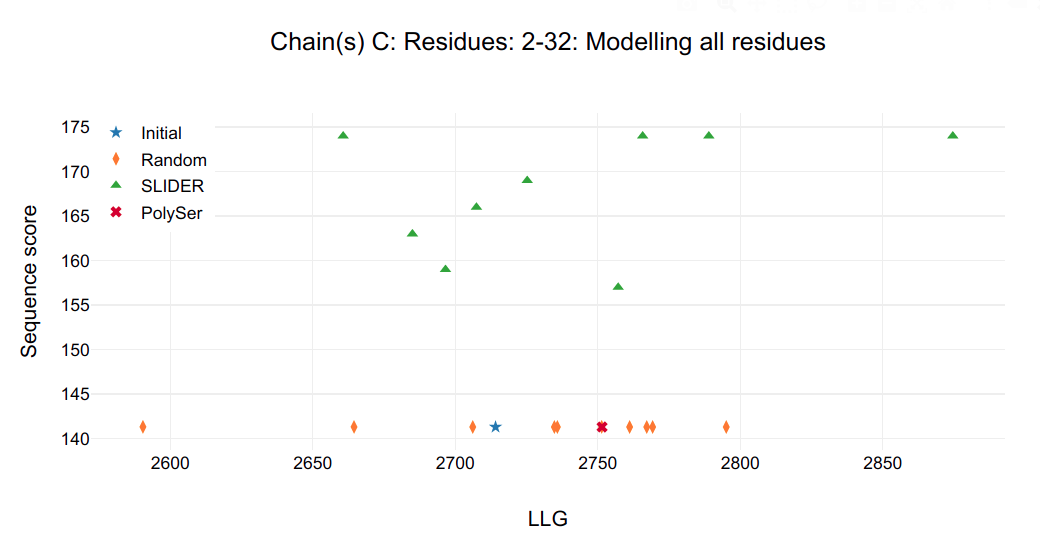
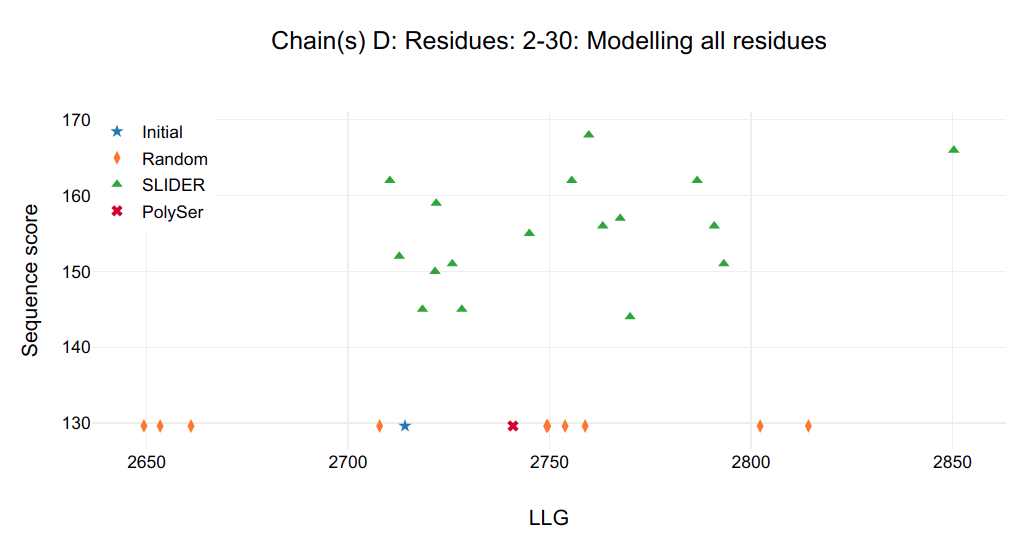
Figure 1.
It is possible to see that a single model in chain C and D are clearly distinguished from all other possibilities, including SLIDER, random and PolySer models.
Looking at the SLIDER log output for chain C (SLIDER/seq_C2-32/Table_refine1_seq_C2-32.log) the best model is seq1_C2-32_ref1 (found in SLIDER/seq_C2-32/1_ref/seq1_C2-32_ref1.pdb) correspondent to sequence hypothesis of residue 34-64 (Fragment 2).
Looking at the SLIDER log output for chain D (SLIDER/seq_D2-30/Table_refine1_seq_D2-30.log) the best model is also seq1_D2-30_ref1 (found in SLIDER/seq_D2-30/1_ref/seq1_D2-30_ref1.pdb) correspondent to sequence hypothesis of residue 37-65 (Fragment 2).

Figure 2.
As the sequence hypotheses of the chain C of one model and chain D of the other overlap in residues 37-64, it is possible to find their non-crystallographic symmetric (NCS) superposing the correspondent residues.
This may be accomplished in COOT:
Open both models in COOT, go to ‘Calculate’ and ‘LSQ Superpose’, select Reference Molecule as seq1_C2-32_ref1.pdb Reference Residue Range 6 to 31 Chain C Reference Molecule as seq1_D2-30_ref1.pdb Reference Residue Range 3 to 28 Chain D
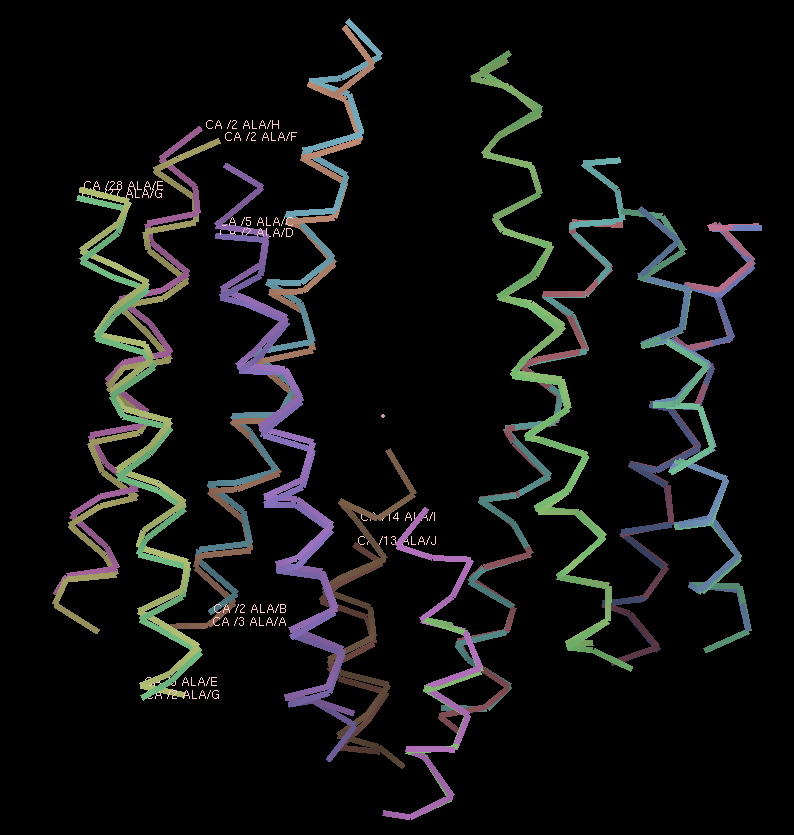
Figure 3.
The resulting superposition is shown in the previous COOT screenshot. Looking at the atom labels, it is possible to see chain A&B, C&D, E&G, F&H and I&J are NCS-related.
Now, let’s shift for the next cycle.
With the NCS found, SLIDER NCS option can be activated to assign sequence to NCS-related chains simultaneously, the increase in the number of assigned residues should improve the hypotheses discrimination for remaning chains. In the current SLIDER code, to use the NCS option, NCS-related chains need to have the same number of residues and same secondary structure. Therefore, residues 2A (residue 2 of monomer A), 2-4C, 30D, 2E, 2-4F, 27F, 2-3H and 2I were removed and helix of J was extended in 4 residues in C-terminal using NCS-related I coordinates. After these alterations, chain C and D with sequence assigned were merged and a new round of SLIDER was set using the keyword “chosen_chains: A,B” and “fixed_residues_modelled: C:5-32 D:2-30”. If you were not able to perform previous steps or you just want to try this new cycle, files are found here.
The output figure shows a single SLIDER model distinguished from rest, it is seq1_A3-34_B2-33 model with sequence hypothesis of 34-65 (Fragment 2 in secondary structure prediction), therefore the other NCS-related chains are discovered: A&B&C&D and E&F&G&H. Applying this new NCS to the other helices, two new helices are found with I&J. Helices E&F&G&H are merged together in order to have same number of residues and should be Fragment 1. Helices I&J are doubled to K&L and should be Fragment 3.
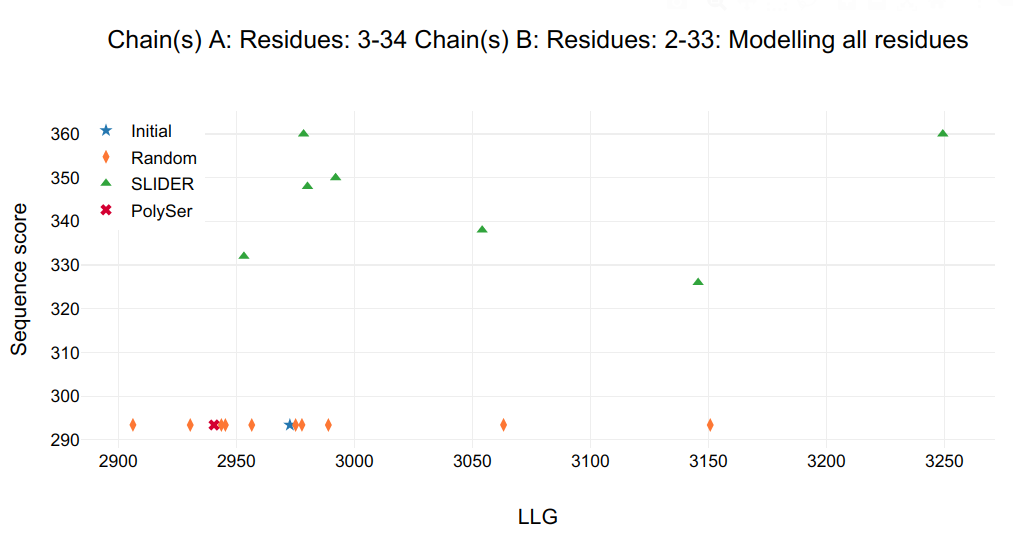
Figure 4.
Next SLIDER run was set for helices E&F&G&H and they should be restricted to Fragment 1, as Fragment 2 was already assigned for all four monomers (chains A-D). In order to accomplish that in SLIDER, the easiest task is to edit PSIPRED prediction file and change the Fragment 2 secondary structure from HELIX (H) to COIL (C). Moreover, the changed keywords in borfile were “chosen_chains: E,F,G,H” and “fixed_residues_modelled: A:1-36 B:1-36 C:1-36 D:1-36”. If you were not able to perform previous steps or you just want to try this new cycle, files are found here.
The graph from the HTML output is shown below and two models are distinguished from rest, they are seq0 (hypothesis 3-29) and seq6 (hypothesis 7-33). Checking the other refinement statistics, Rfactors are the same, but Rfree is 45.2% for seq0 and 46.0% for seq6. Moreover, another interesting strategy to distinguish possibilities, it is looking at the electron density of the initial map with no side chains assigned, and therefore no bias for side chains, and its correspondence to the possible models. With this strategy, the most characterizing feature is the side chain of Arg14 of seq0 (carbons colored in yellow in right hand side in below Figure) bound through salt bridges to Glu55, whereas for seq6 there is the improbable hydrophobic Ile18 (carbons colored in magenta). The maps from refinement of seq0 and seq6 may be compared, but they will have great deal of bias in the assigned side chains, therefore, looking at regions with no side atoms, such as for helices in chains I&J&K&L could give some insights, but for this case, they are pretty much alike. Another strategy is looking if hydrophobic residues are buried and hydrophilic are exposed.
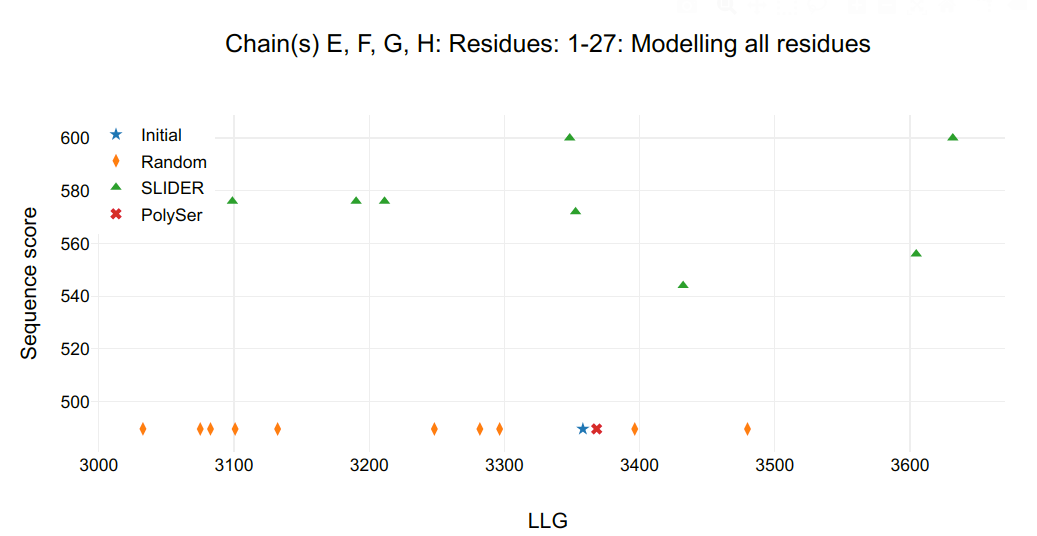
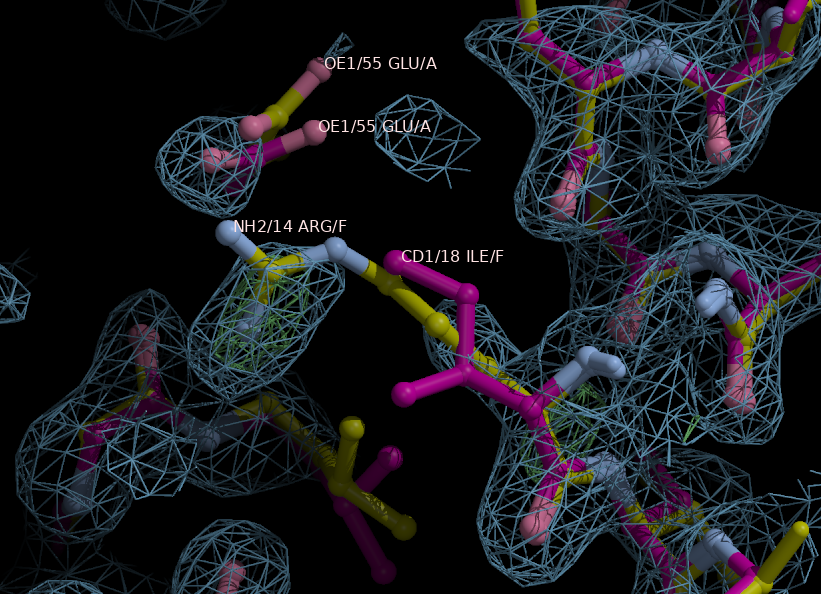
Figure 5.
Going further in sequence assignment, there is the remaining helices in chains I&J&K&L correspondent to Fragment 3. PSIPRED secondary structure of Fragment 1 and 2 were changed to C and a new round of SLIDER was ran using “fixed_residues_modelled: A:1-32 B:1-32 C:4-31 D:3-31 E:1-27 F:1-27 G:1-27 H:1-27” and “chosen_chains: I,J,K,L”.
Unfortunately, a random sequence model had the best LLG score, therefore we decided to try another refinement program, buster (“keyword: refinement_program: buster”). If you were not able to perform previous steps or you just want to try this new cycle, files are found here. Fortunately, the seq0 model, correspondent to hypothesis 72-87, is well distinguished from rest. Therefore, the remaining modeling steps, such as connecting loops, fixing rotamers and adding solvent molecules, are easily done by hand.
References
-
SEQUENCE SLIDER: expanding polyalanine fragments for phasing with multiple side-chain hypotheses.
Borges, R. J., Meindl, K., Triviño, J., Sammito, M., Medina, A., Millán, C., Alcorlo, M., Hermoso, J. A., Fontes, M., and Usón, I.
Acta Cryst. D76, 221-237 (2020) (doi:10.1107/S2059798320000339)
-
ARCIMBOLDO on coiled coils.
Caballero, I., Sammito, M., Millan, C., Lebedev, A., Soler, N. and Uson, I.
Acta Cryst. D74, 194-204 (2018) (doi:10.1107/S1399004715010846)
-
The Effectors and Sensory Sites of Formaldehyde-responsive Regulator FrmR and Metal-sensing Variant.
Osman, D., Piergentili, C., Chen, J., Sayer, L. N., Usón, I., Huggins, T. G., Robinson, N. J., and Pohl, E.
The Journal of biological chemistry, 291(37), 19502–19516 (2016) (doi.org/10.1074/jbc.M116.745174)