SLIDER Remote-Homolog tutorial
Aims of the tutorial: SEQUENCE SLIDER in Remote-Homolog Mode
To illustrate SEQUENCE SLIDER in Remote-Homolog Mode, the structure lipase/acylhydrolase from Enterococcus faecalis was chosen (Borges et al. 2020). It contains a Rossmann fold with 195 residues and its data (PDB id: 1yzf) diffracts to 1.9 Å resolution in P3221 space group (Midwest Center for Structural Genomics, unpublished work). It is possible to obtain its partial solution using ARCIMBOLDO_SHREDDER (Millán et al. 2018) using a homolog from Pseudoalteromonas sp. (PDB id: 3ph4; Jung et al., 2011) that shares 22% identity and shows an r.m.s.d. (root-mean-square deviation) of 2.3 Å over 161 Cα atoms of matching secondary structure. The partial solution is characterized by 81 residues divided into three helices and four strands and by LLG, TFZ and CC of 56, 10.4 and 13.4%, respectively.
Step by Step tutorial
Input
We will need:
All required files can be downloaded here.
The configuration .bor file with the parameters for an SEQUENCE SLIDER run, which is defined as follows:
[CONNECTION]: distribute_computing: multiprocessing [GENERAL]: working_directory: /home/user/SLIDER-RHM/1yzf mtz_path: /home/user/SLIDER-RHM/1yzf/1yzf.mtz hkl_path: /home/user/SLIDER-RHM/1yzf/1yzf.hkl pdb_path: /home/user/SLIDER-RHM/1yzf/ARC_SHREDDER_partial_solution_3hp4.pdb [SLIDER] align_path: /home/user/SLIDER-RHM/1yzf/hhpred-1yzf-3hp4.pir molecular_weight: 21470 number_of_component: 1 f_label: FOBS sigf_label: SIGFOBS rfree_label: R-free-flags refinement_program: phenix.refine PhenixRefineParameters = strategy=individual_sites+individual_adp+individual_sites_real_space+rigid_body ramachandran_restraints=True main.number_of_macro_cycles=5 write_eff_file=false write_geo_file=false write_def_file=false optimize_xyz_weight=False optimize_adp_weight=False secondary_structure.enabled=True export_final_f_model=true simulated_annealing=true sliding_tolerance: 1 seq_pushed_refinement: 10 RandomModels: 10 shelxe_line: -m15 -a25 -s0.5 -v0 -t1 -q -y1.90 -f -B2 -x3 number_shelxe_trials: 10 [LOCAL] path_local_shelxe: /usr/local/ccp4-7.1/bin/shelxe path_local_phenix.refine: /usr/local/phenix-1.18.2-3874/build/bin/phenix.refine path_local_sprout: /usr/local/sprout_linux path_local_edstats: /usr/local/ccp4-7.1/bin/edstats
Execution
Interactively:
SLIDER.py 1yzf.bor
Output
In the HTML, the first figure shows the statistics of sequence hypotheses after refinement:
- in the title, the chain followed by its respective residues whose side chain are being assigned.
- in the Y axis, sequence score, which describes the concordance of the sequence hypothesis in respect to the alignment.
- in X axis, log-likelihood gain (LLG) calculated by PHASER.
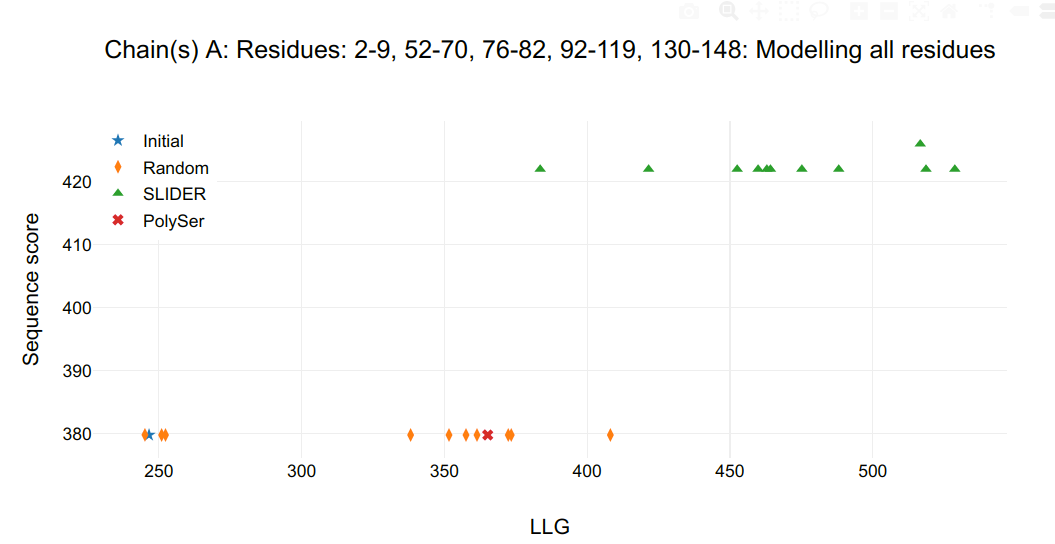
Figure 1. Refinement statistics.
The second figure shows the statistics of sequence hypotheses after expansion:
- in the title, the chain followed by its respective residues whose side chain are being assigned.
- in the Y axis, correlation coefficient (CC) calculated by SHELXE.
- in X axis, log-likelihood gain (LLG) calculated by PHASER.
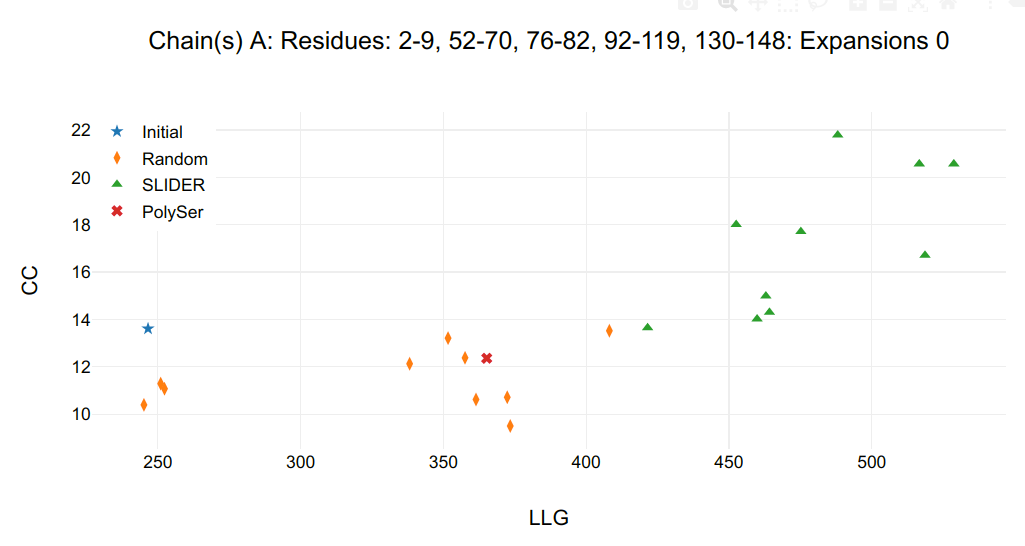
Figure 2. Expansion statistics.
If the expand_from_map option is set to True, expansions from coordinates are numbered 0 and shown in 2nd plot and from maps, 1, shown in third plot. The FOMs of the initial model (star in graph) gives a baseline in respect to the models receiving side chains (triangles in graph). If random sequences (diamonds in graph) are assigned to the initial model, their FOM gives a base line of true negative, even though there will be some correctness in main chain atoms and some correspondence in side chain atoms.
References
-
SEQUENCE SLIDER: expanding polyalanine fragments for phasing with multiple side-chain hypotheses.
Borges, R. J., Meindl, K., Triviño, J., Sammito, M., Medina, A., Millán, C., Alcorlo, M., Hermoso, J. A., Fontes, M., and Usón, I.
Acta Cryst. D76, 221-237 (2020) (doi:10.1107/S2059798320000339)
-
Exploiting distant homologues for phasing through the generation of compact fragments, local fold refinement and partial solution combination.
Millán, C. , Sammito, M. D., McCoy, A. J., Nascimento, A. F., Petrillo, G., Oeffner, R. D., Domínguez-Gil, T. , Hermoso, J. A., Read, R. J. and Usón, I.
Acta Cryst. D74: 290-304 (2018) (doi:10.1107/S2059798318001365)