ARCIMBOLDO_LOW tutorial
Crystallography at low-resolution must determine the atomic model from less experimental observations, which is challenging in the absence of a model. In addition, model bias is the more severe when independent experimental data are scarce.
Our methods solve the phase problem by combining the location of accurate model fragments using Phaser, with density modification and interpretation of the resulting maps using SHELXE. From a partial, correct structure the density modification process and the stereochemical constraints draw the rest of the structure, validating the result. This same principle is now exploited at low-resolution.
Crystallography provides an accurate experimental atomic structure but due to the phase problem, errors in the model bias the determination. At low-resolution, it needs to be established whether the final model is experimental or the data could not disprove the initial, virtually complete model. ARCIMBOLDO, originally an ab initio phasing method, phases challenging structures at low-resolution and introduces the concept of verification, to assess the solution and the capacity of the data to establish it.
Coiled-coils are important, ubiquitous structures but notoriously difficult to phase and to predict. Both correct solutions and incorrect ones are poorly discriminated by the crystallographic figures of merit as long as helices are correctly oriented. We incorporate coiled-coil verification, designed to set up competing, incompatible structural hypotheses to probe both the results and establish the power of the data to discriminate them. SHELXE tracing at low-resolution has been enhanced, maintaining its local character but extending the environment assessment. For non-helical structures, verification is demonstrated in the fragment location process. Relying on verification, we have extended the use of the ARCIMBOLDO software up to 4 Å.
Verification is based on the idea that whereas there are many ways a solution may be incorrect, the true solution must be unique. In practice, we introduce realistic perturbations into the most probable solutions produced, then discrimination among the perturbed solutions and the ones presenting best figures of merit allows us to estimate the correctness of the solution produced. If incompatible solutions are not differentiated by their figures of merit, the solution will remain inconclusive. In ARCIMBOLDO_LITE perturbations generated include a random translation and reversed helices, while in ARCIMBOLDO_SHREDDER only a random translation is performed as we have not seen the case where tracing was reverting parts of the model.
Aims of the tutorial
This tutorial shows how to launch the coiled_coil mode1,2 and predicted_model mode3 implemented in ARCIMBOLDO_SHREDDER4 in order to solve a 78 amino acids structure at 3.3 Å using a prediction obtained with AlphaFold 25 and analyse the output of the program. This example uses the ARCIMBOLDO_SHREDDER version released on July 2024 through CCP46 and our website. This run was performed in a workstation with 8 cores and took less than 3 hours to complete. This structure can also be solved with ARCIMBOLDO_LITE7 in its coiled_coil mode: see tutorial here.
Data tutorial
Experimental details
Test data for our tutorial is the crystal structure of the VBP Leucine Zipper with bound arylstibonic acid. This is a human transcription factor in complex with an inhibitor. The structure, composed by 2 ɑ-helices wrapped around each other to form a coiled coil domain, is deposited in the Protein Data Bank8 under the PDB code 4U5T9.
Crystallographic details are summarized in the following table:
| PDB ID | 4U57 |
| Space group | P61 |
| Unit cell (a, b, c) (Å) | 68.84, 68.84, 77.23 |
| Resolution (Å) | 3.3 |
| Residues in asymmetric unit | 74 |
| Molecular weight | 9340 |

Figure 1. Cartoon representation of PDB entry 4U5T. This figure was prepared using PyMOL10.
Step by Step tutorial
For this tutorial we will need the reflection file in two formats (.mtz and .hkl) and the prediction that will be used as a model for the molecular replacement search (ranked_0.pdb). All required files can be downloaded here. After downloading ARCIMBOLDO_SHREDDER Spheres (see instructions here), you are ready to follow this tutorial.
Required input
We need 4 files in order to run ARCIMBOLDO_SHREDDER Spheres:
Configuration .bor file:
The configuration file looks like follows:
[CONNECTION]: distribute_computing: multiprocessing setup_file: /path/to/setup.bor [GENERAL]: working_directory: /path/to/working_directory mtz_path: %(working_directory)s/4u5t.mtz hkl_path: %(working_directory)s/4u5t.hkl [ARCIMBOLDO] name_job: 4u5t molecular_weight: 9340 number_of_component: 1 f_label: FOBS sigf_label: SIGFOBS number_of_component: 1 model_file: /absolute/path/to/ranked_0.pdb coiled_coil: true predicted_model: true [LOCAL] path_local_phaser: /path/to/phaser path_local_shelxe: /path/to/shelxe
The [CONNECTION] section specifies the computing system used and the file containing the general configuration. The job can be run on a single machine (in multiprocessing) or in a local or remote grid of computers. If you want to run the job in multiprocessing you just have to change in the .bor file that you downloaded, the paths of the working directory, Phaser11 and SHELXE12 (in multiprocessing the setup_file is not required).
The [GENERAL] section contains the .mtz and .hkl file paths as well as the working directory where results will be written.
In the [ARCIMBOLDO] section all parameterization for the job is specified:
It is important to activate the predicted_model mode that entails:
It is important to activate the coiled_coil mode that entails:
Finally, in the [LOCAL] section, the paths to Phaser and SHELXE are set. This is only required if you are in multiprocessing mode (run on a single computer), otherwise the paths must be specified in the configuration setup.bor file.
Execution
You can run the program interactively, having the output displayed on the screen or do it in background, redirecting an input file and passing the output to a .log file.
1. Interactively:
ARCIMBOLDO_SHREDDER 4u5t.bor
2. In background:
nohup ARCIMBOLDO_SHREDDER 4u5t.bor >& logfile.log;
Output and Results
In the directory where you launched ARCIMBOLDO_SHREDDER, you will find a directory called models containing a library of pdbs. Around each Calpha in the template, models are cut in a spatial way, producing a set of non-redundant, overlapping, compact models. In the default mode, they are also annotated in different chains in order to decompose them and perform gyre and gimble refinement, aiming to give more degrees of freedom and obtain a more accurate model. Also you will find an html output with a link to the html output of the library search. The first section echoes all parameters used for the run, so that defaults are listed along with values for the parameters set through the .bor file. This allows to reproduce the run even if defaults may change in future versions.
The directory called ARCIMBOLDO_BORGES contains the output of the ARCIMBOLDO_BORGES16 run using the library in models. The html output of the library search is found here. The next section displays a graph and a table summarizing the rotation clustering step as in ARCIMBOLDO_BORGES. A sortable table follows, summarizing the results for all Phaser and SHELXE steps, including top and average figures of merit for each rotation cluster that has been evaluated.
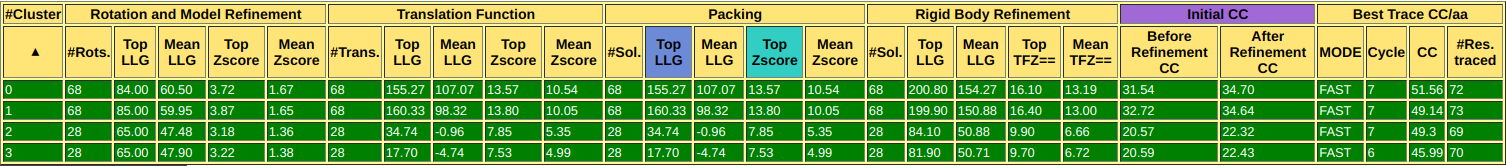
Figure 2. Interactive table with the figures of merit from Phaser and SHELXE for each rotation cluster.
After the table, you can find the backtrace and figures of merit for the best solution, in this case the structure is solved with a CC of 51.56% and 72 residues traced. Also, there are links to access the best scoring solution: the .pdb of the traced structure and its map in .phs format.
Figure 3. Backtracking of the model that leads to a correct solution.
After backtracking, a verification graph indicates that the structure is solved. It is crucial to check this verification result to ensure the reliability of the generated structural hypothesis. In this case, the best solution is clearly distinguishable from a random one.
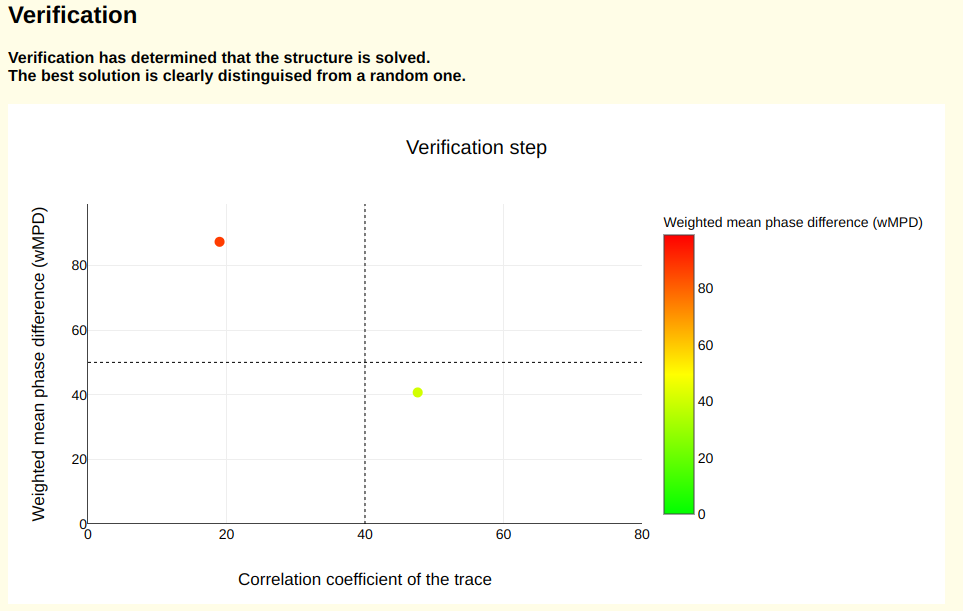
Figure 4. Verification step graph.
Finally, the last section of this html containing a full configuration file will be reported, showing the parameterization used to launch the library of fragments. It includes all parameters, the ones provided by the user as well as the defaults that remained unchanged, and the log file of the run.
References
-
ARCIMBOLDO on coiled coils.
Caballero, I., Sammito, M., Millan, C., Lebedev, A., Soler, N. and Uson, I.
Acta Cryst. D74, 194-204 (2018) (doi:10.1107/S2059798317017582)
-
ARCIMBOLDO at low-resolution: Verification for coiled-coils and globular proteins.
Caballero I., Castellví A., Triviño J., Jiménez E., Soler N., Borges R. and Usón I.
Protein Sci. 33(9), e5136 (2024) (doi:10.1002/pro.5136)
-
Verification: model-free phasing with enhanced predicted models in ARCIMBOLDO_SHREDDER.
Medina, A., Jiménez, E., Caballero, I., Castellví, A., Triviño Valls, J., Alcorlo, M., Molina, R., Hermoso, J. A., Sammito, M. D., Borges, R. & Usón, I.
Acta Cryst. D78, 1283–1293 (2022) (doi:10.1107/S2059798322009706)
-
Exploiting distant homologues for phasing through the generation of compact fragments, local fold refinement and partial solution combination.
Millán, C., Sammito, M. D., McCoy, A. J., Nascimento, A. F., Petrillo, G., Oeffner, R. D., Domínguez-Gil, T., Hermoso, J. A., Read, R. J. and Usón, I.
Acta Cryst. D74, 290-304 (2018) (doi:10.1107/S2059798318001365)
-
Highly accurate protein structure prediction with AlphaFold.
Jumper, J., Evans, R., Pritzel, et al.
Nature 596, 583–589 (2021) (doi:10.1038/s41586-021-03819-2)
-
The CCP4 suite: integrative software for macromolecular crystallography.
Aguirre, J. et al.
Acta Cryst. D79, 449-461 (2023) (doi:10.1107/S2059798323003595)
-
ARCIMBOLDO_LITE: single-workstation implementation and use.
Sammito, M., Millán, C., Frieske, D., Rodríguez-Freire, E., Borges, R. J. and Usón, I.
Acta Cryst. D71, 1921-30 (2015) (doi:10.1107/S1399004715010846)
-
RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy.
Burley, S. et al.
Nucleic Acids Research, 47(D1), D464–D474 (2018) (doi:10.1093/nar/gky1004)
-
P6981, An Arylstibonic Acid, Is a Novel Low Nanomolar Inhibitor of cAMP Response Element-Binding Protein Binding to DNA.
Zhao, J., Stagno, J.R., Varticovski, L., Nimako, E., Rishi, V., McKinnon, K., Akee, R., Shoemaker, R.H., Ji, X., Vinson, C.
Molecular Pharmacology, 82 (5) 814-823 (2012) (doi:10.1124/mol.112.080820)
-
The PyMOL Molecular Graphics System.
Version 1.5.0.4 Schrödinger, LLC. (https://pymol.org)
-
Phaser crystallographic software.
McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. and Read, R. J.
J. Appl. Crystallogr. D40, 658-674 (2007) (doi:10.1107/S0021889807021206)
-
Modes and model building in SHELXE.
Usón, I. & Sheldrick, G. M.
Acta Cryst. D80 (Pt 1), 4–15 (2024) (doi:10.1107/S2059798323010082)
-
On the application of the expected log-likelihood gain to decision making in molecular replacement.
Oeffner, R D, Afonine, P. V, Millán, C., Sammito, M., Usón, I., Read, R. J., & McCoy, A.
J. Acta Cryst. D74 (Pt 4), 245–255 (2018) (doi:10.1107/S2059798318004357)
-
Gyre and gimble: a maximum-likelihood replacement for Patterson correlation refinement.
McCoy, A. J., Oeffner, R. D., Millan, C., Sammito, M., Uson, I. and Read, R. J.
Acta Cryst. D74, 279-289 (doi:10.1107/S2059798318001353)
-
Overview of the CCP4 suite and current developments.
Winn, M.D. et al.
Acta Cryst. D67, 235-242 (2011) (doi: 10.1107/S0907444910045749)
-
Exploiting distant homologues for phasing through the generation of compact fragments, local fold refinement and partial solution combination.
Millán, C. , Sammito, M. D., McCoy, A. J., Nascimento, A. F., Petrillo, G., Oeffner, R. D., Domínguez-Gil, T. , Hermoso, J. A., Read, R. J. and Usón, I.
Acta Cryst. D74: 290-304 (2018) (doi:10.1107/S2059798318001365)