Manual
The following documentation describes the current version of the programs. We encourage upgrade to the latest version as bugs have been corrected and new features are available.
ARCIMBOLDO_LITE manual
Overview
Mainly helical proteins or those having helices as a prominent feature are particularly suited for ARCIMBOLDO phasing. Therefore, polyalanine model helices of selected lenght are internaly provided. Any other model can be externally provided through a PDB file.
Input
- An mtz file containing the reflection data.
- A SHELX reflection file hkl containing the reflection data.
- The configuration .bor file with the parameters for an ARCIMBOLDO run, which is defined as follows:
[CONNECTION]: #Values for the following keyword are mutually excluding distribute_computing: multiprocessing #distribute_computing: local_grid #distribute_computing: remote_grid #default is to search your rsa private keyfile into ~/.ssh/id_rsa #remote_frontend_passkey: #setup_bor_path: [GENERAL]: working_directory: mtz_path: hkl_path: #ent_path: [ARCIMBOLDO-LITE] name_job: molecular_weight: number_of_component: i_label: sigi_label: #f_label: #sigf_label: fragment_to_search: 2 helix_length: search_inverted_helix: False search_inverted_helix_from_fragment: -1 top_inverted_solution_per_cluster: 1000 #model_file: #fixed_models_directory: coiled_coil: False rmsd: 0.2 resolution_rotation: 1.0 sampling_rotation: -1 resolution_translation: 1.0 sampling_translation: -1 resolution_refinement: 1.0 sampling_refinement: -1 exclude_llg: 0 exclude_zscore: 0 use_packing: True pack_clashes: 0 pack_distance: 3.0 pack_tra: False occ: False tncs: True vrms: False update_rmsd: False bfac: False solution_sorting_scheme: AUTO #solution_sorting_scheme: LLG #solution_sorting_scheme: ZSCORE #solution_sorting_scheme: INITCC #solution_sorting_scheme: COMBINED #shelxe_line: nice: 0 save PHS: False usepdo: False topfrf_1: 1000 topftf_1: 150 toppack_1: 10000 toprnp_1: 1000 topexp_1: 60 topfrf_n: 200 topftf_n: 150 toppack_n: 10000 toprnp_n: 150 topexp_n: 60 force_core: -1 force_exp: False #The following section is only required in multiprocessing mode [LOCAL] path_local_phaser: path_local_shelxe:
[CONNECTION]
[distribute_computing]: multiprocessing or local_grid or remote_grid. Default is multiprocessing on a single machine. If a grid is used, the next variables should be defined.
[setup_bor_path]: path to the configuration file for program setup.
[remote_frontend_passkey]: False. If you want to use your personal id_rsa key that is not stored in the default path ~/.ssh/id_rsa, then put the full path here.
[GENERAL]
These configuration variables refer to things that need to be setup in either of the modes.
[mtz_path]: path to the mtz file with reflection data.
[hkl_path]: path to the hkl file with reflection data.
[working_directory]: absolute path to working directory.
[ARCIMBOLDO-LITE]
Next variables are mandatory and should be input by the user.
[name_job]: string, should be unique to the run and have a max length of 20 non-special characters except "_" (underscore).
[molecular_weight]: it will be used by PHASER to calculate the composition in the ASU, assuming that protein/nucleic acid have the average distribution of aminoacids and bases.
[number_of_component]: number of copies of protein/nucleic acid defined by the molecular weigth.
Latest Phaser versions are able to work directly with intensities, because new likelihood targets and functions have been defined. We strongly recommend to use this feature if possible. For that purpose, use the keywords i_label and sigi_label in the bor file to indicate the columns from the mtz.
[i_label]: label for the intensities in the mtz file.
[sigi_label]: label for the standard deviation of the intensities in the mtz file.
[f_label]: label for the amplitudes in the mtz file.
[sigf_label]: label for the standard deviation of the amplitudes in the mtz file.
A search model can be defined in four different ways:
[helix_length]: number of residues. To search an ideal helix of the specified length the number of times you have defined in [fragment_to_search]
or
[model_file]: /path/to/model_file.pdb. To search the specified model the number of times defined in [fragment_to_search]
or
[helix_length_n]: number of residues. To search for as many helices of different sizes as [fragment_to_search] (e.g. [helix_length_1], [helix_length_2], [helix_length_3], etc.).
or
[model_file_n]: name_model_file. To provide as many different models as [fragment_to_search] (e.g. [model_file_1], [model_file_2], [model_file_3], etc.).
The following parameters may be modified but are not mandatory because defaults are provided (bold) and are presented in two groups (basic and advanced):
Basic
[fragment_to_search]: 2. Total number of models that you will search for.
[shelxe_line]: String, command line for SHELXE. If unset, sensible default values depending on resolution will be used.
[rmsd]: 0.2.The expected RMS deviation of the coordinates to the "real" structure, used by PHASER. As fragments are small, we can consider them very similar. In any case the range accepted is 0.2 - 2.4.
Advanced
[coiled_coil]:False or True. If True, automatic parameterisation for coiled coil cases will be used.
[search_inverted_helix]: False or True. If True, after rotation search, helices are tested both in their current orientation and in the alternative 180º.
[search_inverted_helix_from_fragment]: -1 . Fragment search cycle from which to apply the inverted helix search. Default is all fragments, if given an integer number, it starts from such fragment.
[top_inverted_solution_per_cluster]: 1000. Number of solutions to test for inversion. Default is the first 1000 rotations per cluster, sorted by LLG.
[fixed_models_directory]: path to a folder containing pdb files. Each of them will be independently used as a fixed fragment before performing the search. [fragment_to_search] must be changed accordingly.
[resolution_rotation]: 1.0
[sampling_rotation]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[resolution_translation]: 1.0
[sampling_translation]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[resolution_refinement]: 1.0
[sampling_refinement]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[exclude_llg]: 0. Threshold to exclude solutions that have an LLG below the value set.
[exclude_zscore]: 0. Threshold to exclude solutions that have a ZSCORE below the value set.
[use_packing]: True or False. If True, PHASER's packing test will be applied to the rototranslated solutions. Otherwise, packing test will be skipped.
[pack_tra]: False or True. If True, the top translation peaks will be tested for packing with the same criteria than the one used afterwards by ARCIMBOLDO. In that way, you will ensure that top solution from translation will survive the packing test.
[pack_clashes]: 0. Number of allowed clashes in the PHASER packing test.
[pack_distance]: 3.0. Threshold for atom contacts in the PHASER packing test.
[occ]: False or True. If True, activates PHASER's occupancy refinement of solutions.
[tncs]: True or False. It enables / disables the PHASER feature for translational non crystallographic symmetry.
[vrms]: False or True. If True, activates PHASER's variance rms refinement.
[bfac]: False or True. If True, activates PHASER's bfactor refinement.
[gimble]: False or True. If True, and in case model_file has been divided in rigid groups by chains, PHASER's gimble refinement of solutions will be performed.
[sigr]: 0.0. Limit in degrees for the rotation in gimble refinement.
[sigt]: 0.0. Limit in Ångström for the translation in gimble refinement.
[solution_sorting_scheme]: AUTO or LLG or ZSCORE or COMBINED. Method to prioritize solutions for shelxe expansion.
[nice]: 0. In multiprocessing mode, it can be used to invoke ARCIMBOLDO_LITE with a particular priority for the CPU time. -20 is the highest priority and 19 is the lowest one.
[savephs]: False or True. If True, the phs map files from the initial correlation coefficient calculation step will be saved.
[usepdo]: False or True. If shelxe -o optimization has been set on the shelxe line, and fragment search includes more than one fragment, on each subsequent search, the trimmed model and not the whole helix will be used as the fixed fragment for searching the next one.
[topfrf_1]: 1000. Limit of rotation solutions at the first fragment search.
[topftf_1]: 150. Limit of translation solutions at the first fragment search.
[toppack_1]: 10000. Limit of packing solutions at the first fragment search.
[toprnp_1]: 1000. Limit of refinement solutions at the first fragment search.
[topexp_1]: 60. Limit of expansion solutions at the first fragment search.
[topfrf_n]: 200. Limit of rotation solutions for the remaining fragments to search.
[topftf_n]: 150. Limit of translation solutions for the remaining fragments to search.
[toppack_n]: 10000. Limit of packing solutions for the remaining fragments to search.
[toprnp_n]: 150. Limit of refinement solutions for the remaining fragments to search.
[topexp_n]: 60. Limit of expansion solutions for the remaining fragments to search.
[force_core]: -1. Default means that in multiprocessing mode, all physical cores minus one will be used for running ARCIMBOLDO. If set to an integer value, it will use that number of cores.
[force_exp]: False or True. f True, it will expand double the number of solutions that the program would normally expand.
[LOCAL]
[path_local_phaser]: path to your local PHASER installation.
[path_local_shelxe]: path to your local SHELXE installation.
Execution
Interactively:
ARCIMBOLDO_LITE input.bor
In background:
When using a grid in which a password is required, it may be given as input in a text file (e.g. password). Otherwhise, just redirecting the output and using & will launch the job in background.ARCIMBOLDO_LITE input.bor < password >& log &
Output
The .html output file created will summarize the figures of merit rendered by PHASER determining the partial structure and SHELXE autotracing of the total structure. If the procedure gave rise to an interpretable map SHELXE could trace, the correlation coefficient between the model and the data should be higher than 30%. If this is the case, the protein was probably solved. Links to the best trace and map can be found on the html output.
Go to top page
ARCIMBOLDO_BORGES manual
Overview:
ARCIMBOLDO_BORGES can phase a structure expected to contain a given small fold using a library of tertiary structure fragments and diffraction data to 2Å.
Input
- An mtz file containing the reflection data.
- A SHELX reflection file hkl containing the reflection data.
- The configuration .bor file with the parameters for ARCIMBOLDO-BORGES, which has the [CONNECTION], [GENERAL] and [LOCAL] sections defined as in an ARCIMBOLDO run, and a particular [ARCIMBOLDO-BORGES] section.
[CONNECTION]: #Values for the following keyword are mutually excluding distribute_computing: multiprocessing #distribute_computing: local_grid #distribute_computing: remote_grid #default is to search your rsa private keyfile into ~/.ssh/id_rsa #remote_frontend_passkey: #setup_bor_path: [GENERAL]: working_directory: mtz_path: hkl_path: ent_path: [ARCIMBOLDO-BORGES] name_job: molecular_weight: number_of_component: i_label: sigi_label: #f_label: #sigf_label: library_path: clusters: all n_clusters: 4 prioritize_phasers: True rmsd: 0.2 resolution_rotation: 1.0 sampling_rotation: -1 resolution_translation: 1.0 sampling_translation: -1 resolution_refinement: 1.0 sampling_refinement: -1 resolution_gyre: 1.0 sampling_gyre: -1 exclude_llg: 0 exclude_zscore: 0 use_packing: True pack_clashes: 0 pack_distance: 3.0 pack_tra: False occ: False prioritize_occ: True tncs: True vrms: False bfac: False gimble: False nma: False sigr: 0.0 sigt: 0.0 gyre_preserve_chains: False rotation_model_refinement: NO GYRE #rotation_model_refinement: BOTH #rotation_model_refinement: GYRE solution_sorting_scheme: AUTO #solution_sorting_scheme: LLG #solution_sorting_scheme: ZSCORE #solution_sorting_scheme: INITCC #solution_sorting_scheme: COMBINED #shelxe_line: #nice: 0 alixe: True alixe_mode: monomer savephs: False extend_with_secondary_structure: False parameters_elongation: 4.8 60 150 #parameters_elongation: 5 150 1 topfrf: 200 topftf: 70 toppack: -1 toprnp: 200 topexp: 60 force_core: -1 force_nsol: -1 force_exp: False [LOCAL] path_local_phaser: path_local_shelxe:
[CONNECTION]
[distribute_computing]: multiprocessing or local_grid or remote_grid. Default is multiprocessing on a single machine. If a grid is used, the next variables should be defined.
[setup_bor_path]: path to the configuration file for program setup.
[remote_frontend_passkey]: False. If you want to use your personal id_rsa key that is not stored in the default path ~/.ssh/id_rsa, then put the full path here.
[GENERAL]
These configuration variables refer to things that need to be setup in either of the modes.
[mtz_path]: path to the mtz file with reflection data.
[hkl_path]: path to the hkl file with reflection data.
[working_directory]: absolute path to working directory.
[ARCIMBOLDO-BORGES]
Next variables are mandatory and should be input by the user.
[name_job]: string, should be unique to the run and have a max length of 20 non-special characters except "_" (underscore).
[number_of_component]: number of copies of protein/nucleic acid defined by the molecular weigth.
[molecular_weight]: it will be used by PHASER to calculate the composition in the ASU, assuming that protein/nucleic acid have the average distribution of aminoacids and bases.
Latest Phaser versions are able to work directly with intensities, because new likelihood targets and functions have been defined. We strongly recommend to use this feature if possible. For that purpose, use the keywords i_label and sigi_label in the bor file to indicate the columns from the mtz.
[i_label]: label for the intensities in the mtz file.
[sigi_label]: label for the standard deviation of the intensities in the mtz file.
[f_label]: label for the amplitudes in the mtz file.
[sigf_label]: label for the standard deviation of the amplitudes in the mtz file.
[library_path]: path to the library folder.
[shelxe_line]: String, command line for SHELXE. If unset, sensible default values depending on resolution will be used.
[clusters]: "all" or number/list of numbers. All will perform sequentially all clusters, otherwise a list of cluster numbers separated by commas may be specified.
The following parameters may be modified but are not mandatory because defaults are provided (bold) and are presented in two groups (basic and advanced):
Basic
[fragment_to_search]: 2. Total number of models that you will search for.
[shelxe_line]: String, command line for SHELXE. If unset, sensible default values depending on resolution will be used.
[rmsd]: 0.2.The expected RMS deviation of the coordinates to the "real" structure, used by PHASER. As fragments are small, we can consider them very similar. In any case the range accepted is 0.2 - 2.4.
Advanced
[n_clusters]: 4. Number of prioritised clusters to evaluate.
[prioritize_phasers]:True or False. If True, all phaser steps are performed for all selected rotation clusters and shelxe expansions are performed at the end.
[coiled_coil]:False or True. If True, automatic parameterisation for coiled coil cases will be used.
[resolution_rotation]: 1.0
[sampling_rotation]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[resolution_translation]: 1.0
[sampling_rotation]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[resolution_refinement]: 1.0
[sampling_refinement]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[resolution_gyre]: 1.0.
[sampling_gyre]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[exclude_llg]: 0. Threshold to exclude solutions that have an LLG below the value set.
[exclude_zscore]: 0. Threshold to exclude solutions that have a ZSCORE below the value set.
[use_packing]: True or False. If True, PHASER's packing test will be applied to the rototranslated solutions. Otherwise, packing test will be skipped.
[pack_tra]: False or True. If True, the top translation peaks will be tested for packing with the same criteria than the one used afterwards by ARCIMBOLDO. In that way, you will ensure that top solution from translation will survive the packing test.
[pack_clashes]: 0. Number of allowed clashes in the PHASER packing test.
[pack_distance]: 3.0. Threshold for atom contacts in the PHASER packing test.
[occ]: False or True. If True, activates PHASER's occupancy refinement of solutions.
[prioritize_occ]: True or False. If True, solutions that have been occupancy-refined will be prioritised for expansion with shelxe.
[tncs]: True or False. It enables / disables the PHASER feature for translational non crystallographic symmetry.
[vrms]: False or True. If True, activates PHASER's variance rms refinement.
[bfac]: False or True. If True, activates PHASER's bfactor refinement.
[gimble]: False or True. If True, and in case model_file has been divided in rigid groups by chains, PHASER's gimble refinement of solutions will be performed.
[nma]: False or True. If True, PHASER's Normal Mode Analysis will be tested after the packing analysis as another approach for solution refinement. The program will compute Initial CC with shelxe for the obtained models and select the improved ones. Please, note that activate this keyword can produce thousands phasers jobs and shelxe to be performed.
[sigr]: 0.0. Limit in degrees for the rotation in gimble refinement.
[sigt]: 0.0. Limit in Ångström for the translation in gimble refinement.
[gyre_preserve_chains]: False or True. If True, the chains present in the input models will be used for gyre and gimble refinement.
[rotation_model_refinement]: NO GYRE or GYRE or BOTH.
[solution_sorting_scheme]: AUTO or LLG or ZSCORE or COMBINED. Method to prioritize solutions for shelxe expansion.
[nice]: 0. In multiprocessing mode, it can be used to invoke ARCIMBOLDO_BORGES with a particular priority for the CPU time. -20 is the highest priority and 19 is the lowest one.
[alixe]: True or False. If True, phase combination will be applied to the density-modified solutions before expansion.
[alixe_mode]: monomer.
[savephs]: False or True. If True, the phs map files from the initial correlation coefficient calculation step will be saved.
[extend_with_secondary_structure]: False or True.
[parameters_elongation]: 4.8 60 150 or 5 150 1
[topfrf]: 200. Limit of rotation solutions.
[topftf]: 70. Limit of translation solutions.
[toppack]: -1. Limit of packing solutions. By default, all surviving solutions are kept.
[toprnp]: 200. Limit of rigid body refined solutions.
[topexp]: 60. Limit of solutions to expand.
[force_core]: -1. Default means that in multiprocessing mode, all physical cores minus one will be used for running ARCIMBOLDO. If set to an integer value, it will use that number of cores.
[force_nsol]: -1. Default chooses automatically the number of solutions to keep based on available hardware. If given an integer number, will keep that number of solutions per step.
[force_exp]: False or True. f True, it will expand double the number of solutions that the program would normally expand.
Execution
Interactively:
ARCIMBOLDO_BORGES input.bor
In background:
When using a grid in which a password is required, it may be given as input in a text file (e.g. password). Otherwhise, just redirecting the output and using & will launch the job in background.ARCIMBOLDO_BORGES input.bor < password >& log &
Output
The .html output file created will summarize the figures of merit rendered by PHASER determining the partial structure and SHELXE autotracing of the total structure. If the procedure gave rise to an interpretable map SHELXE could trace, the correlation coefficient between the model and the data should be higher than 30%. If this is the case, the protein was probably solved. Links to the best trace and map can be found on the html output.
Go to top page
ARCIMBOLDO-SHREDDER manual
Overview:
A model expected to have some structural similarity to the target protein is the template to generate a set of search fragments with SHREDDER for use in ARCIMBOLDO. The experimental data are used to guide removal of the more incorrect parts of the model. Various approaches for model selection are covered in the following sections.
Input
- An mtz file containing the reflection data.
- A SHELX reflection file hkl containing the reflection data.
- The configuration .bor file with the parameters for ARCIMBOLDO-SHREDDER, which has the [CONNECTION], [GENERAL] and [LOCAL] sections defined as in an ARCIMBOLDO run, and a particular [ARCIMBOLDO-SHREDDER] section.
[CONNECTION]: #Values for the following keyword are mutually excluding distribute_computing: multiprocessing #distribute_computing: local_grid #distribute_computing: remote_grid #default is to search your rsa private keyfile into ~/.ssh/id_rsa #remote_frontend_passkey: #setup_bor_path: [GENERAL]: working_directory: mtz_path: hkl_path: ent_path: [ARCIMBOLDO-SHREDDER]: name_job: molecular_weight: number_of_component: i_label: sigi_label: #f_label: #sigf_label: number_cycles_model_refinement: 2 model_file: fragment_to_search: 1 trim_to_polyala: True maintaincys: False clusters: all n_clusters: 4 prioritize_phasers: True rmsd_shredder: 1.2 rmsd_arcimboldo: 0.8 resolution_rotation_shredder: 1.0 sampling_rotation_shredder: -1 resolution_rotation_arcimboldo: 1.0 sampling_rotation_arcimboldo: -1 resolution_translation: 1.0 sampling_translation: -1 resolution_refinement: 1.0 sampling_refinement: -1 resolution_gyre: 1.0 sampling_gyre: -1 exclude_llg: 0 exclude_zscore: 0 use_packing: True pack_clashes: 3 pack_distance: 3.0 pack_tra: False occ: False prioritize_occ: True tncs: True vrms: False update_rmsd: False bfac: False bfacnorm: True gimble: False sigr: 0.0 sigt: 0.0 gyre_preserve_chains: False rotation_model_refinement: BOTH #rotation_model_refinement: NOGYRE #rotation_model_refinement: GYRE solution_sorting_scheme: AUTO #solution_sorting_scheme: LLG #solution_sorting_scheme: ZSCORE #solution_sorting_scheme: INITCC #solution_sorting_scheme: COMBINED #shelxe_line: #nice: 0 alixe: True alixe_mode: monomer savephs: False shred_method: sequential #shred_method: spherical shred_range: 4 20 1 omit all topfrf: 200 topftf: 70 toppack: -1 toprnp: 200 topexp: 40 force_core: -1 force_nsol: -1 force_exp: False [LOCAL] path_local_phaser: path_local_shelxe:
The following variables are mandatory and should be input by the user.
[name_job]: string, should be unique to the run and have a max length of 20 non-special characters except "_" (underscore).
[molecular_weight]: used by PHASER to calculate the composition of the ASU.
[number_of_component]: number of copies of protein/nucleic acid defined by the molecular weigth.
Latest Phaser versions are able to work directly with intensities, because new likelihood targets and functions have been defined. We strongly recommend to use this feature if possible. For that purpose, use the keywords i_label and sigi_label in the bor file to indicate the columns from the mtz.
[i_label]: label for the intensities in the mtz file.
[sigi_label]: label for the standard deviation of the intensities in the mtz file.
[f_label]: label for the amplitudes in the mtz file.
[sigf_label]: label for the standard deviation of the amplitudes in the mtz file.
The following variables can be modified but are not mandatory because they have sensible preset defaults (bold). They are presented in two groups (basic and advanced):
Basic
[fragment_to_search]: 1. Number of model copies to be located in the asymmetric unit.
[shelxe_line]: String, command line for SHELXE. If unset, sensible default values depending on resolution will be used.
[rmsd_shredder]: 1.2. The expected RMS deviation in &A of the coordinates to the "real" structure, used by PHASER in the evaluation of the shredded models. As fragments are not very similar overall, this value is intentionally underestimated. In any case the accepted range is 0.2 - 2.4.
[rmsd_arcimboldo]: 1.0. The expected RMS deviation in &A of the coordinates to the target structure, used by PHASER in the ARCIMBOLDO runs from the best models. As models have been both reduced and optimized, a lower figure than for the SHREDDER procedure is appropriate. In any case the range accepted is 0.2 - 2.4.
[shred_method]: sequential or spherical.
If the selected method is the sequential:
[shred_range]: 4 20 1 omit. Range of sizes for the shreds (4-20), step between starting residues (1). If keyword omit is chosen, shreds will be eliminated from the structure. Else, with keyword fragment, the shreds will be extracted as search fragments.
If the selected method is the spherical:
sphere_definition: default 1 remove_coil 7 4 0.45 0.2. Size of the model (default based on eLLG target), step between starting residues (1), remove_coil o maintain_coil, minimum size of alpha helices in template, minimum size of beta strands in template, minimum threshold for helix annotation, minimum threshold for beta annotation.
ellg_target: 60. Target eLLG to guide selection of the models size for the given rmsd.
Advanced
[number_cycles_model_refinement]: 2. Number of gyre refinement cycles to perform on the models.
[trim_to_polyala]: True or False. If True, template model will be trimmed to polyalanine.
[maintaincys]: False or True. If True, when trimmed to polyalanine, cysteine residues will keep their sidechain .
[clusters]: "all" or number/list of numbers. All will perform sequentially all clusters, otherwise a list of cluster numbers separated by commas may be specified.
[n_clusters]: 4. Number of prioritised clusters to evaluate.
[prioritize_phasers]:True or False. If True, all phaser steps are performed for all selected rotation clusters and shelxe expansions are performed at the end.
[resolution_rotation_shredder]: 1.0. The resolution limit for the FRF from which to cluster rotations in the SHREDDER runs. Default is using 1.0 or full resolution if data are not available up to 1.0.
[sampling_rotation_shredder]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[resolution_rotation_arcimboldo]: 1.0. Is the resolution to perform the FRF in order to cluster rotations in the ARCIMBOLDO_LITE or ARCIMBOLDO_BORGES runs. Default is using 1.0 or full resolution if data are not available up to 1.0.
[sampling_rotation_arcimboldo]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[resolution_translation]: 1.0. Is the resolution limit for the translation function in the ARCIMBOLDO_LITE or ARCIMBOLDO_BORGES runs. Default is using 1.0 or full resolution if data are not available up to 1.0.
[sampling_translation]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[resolution_refinement]: 1.0. Is the resolution limit for the refinement in the ARCIMBOLDO_LITE or ARCIMBOLDO_BORGES runs. Default is using 1.0 or full resolution if data are not available up to 1.0.
[sampling_refinement]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[resolution_gyre]: 1.0. Is the resolution limit for the gyre refinement in the ARCIMBOLDO_BORGES runs. Default is using 1.0 or full resolution if data are not available up to 1.0.
[sampling_gyre]: -1. Default PHASER sampling, which is dynamically calculated considering model and data.
[exclude_llg]: 0. Threshold to exclude solutions that have an LLG below the value set.
[exclude_zscore]: 0. Threshold to exclude solutions that have a ZSCORE below the value set.
[use_packing]: True or False. If True, PHASER's packing test will be applied to the rototranslated solutions. Otherwise, packing test will be skipped.
[pack_tra]: False or True. If True, the top translation peaks will be tested for packing with the same criteria than the one used afterwards by ARCIMBOLDO. In that way, you will ensure that top solution from translation will survive the packing test.
[pack_clashes]: 3. Percentage of allowed clashes in the PHASER packing test.
[pack_distance]: 3.0. Threshold for atom contacts in the PHASER packing test.
[occ]: False or True. If True, activates PHASER's occupancy refinement of solutions.
[prioritize_occ]: True or False. If True, solutions that have been occupancy-refined will be prioritised for expansion with shelxe.
[tncs]: True or False. It enables / disables the PHASER feature for translational non crystallographic symmetry.
[vrms]: False or True. If True, activates PHASER's variance rms refinement.
[bfac]: False or True. If True, activates PHASER's bfactor refinement.
[bfacnorm]: True or False. If True, bfactors in the input template are set to a constant value.
[gimble]: False or True. If True, and in case model_file has been divided in rigid groups by chains, PHASER's gimble refinement of solutions will be performed.
[sigr]: 0.0. Limit in degrees for the rotation in gimble refinement.
[sigt]: 0.0. Limit in Ångström for the translation in gimble refinement.
[gyre_preserve_chains]: False or True. If True, the chains present in the input models will be used for gyre and gimble refinement.
[rotation_model_refinement]: NOGYRE for sequential mode, BOTH for spherical mode. Can also be GYRE.
[solution_sorting_scheme]: AUTO or LLG or ZSCORE or COMBINED. Method to prioritize solutions for shelxe expansion.
[nice]: 0. In multiprocessing mode, it can be used to invoke ARCIMBOLDO_SHREDDER with a particular priority for the CPU time. -20 is the highest priority and 19 is the lowest one.
[alixe]: True or False. If True, phase combination will be applied to the density-modified solutions before expansion.
[alixe_mode]: monomer.
[savephs]: False or True. If True, the phs map files from the initial correlation coefficient calculation step will be saved.
[topfrf]: 200. Limit of rotation solutions.
[topftf]: 70. Limit of translation solutions.
[toppack]: -1. Limit of packing solutions. Default is all the surviving ones.
[toprnp]: 200. Limit of rigid body refined solutions.
[topexp]: 40. Limit of solutions to expand.
[force_core]: -1. Default means that in multiprocessing mode, all physical cores minus one will be used for running ARCIMBOLDO. If set to an integer value, it will use that number of cores.
[force_nsol]: -1. Default chooses automatically the number of solutions to keep based on available hardware. If given an integer number, will keep that number of solutions per step.
[force_exp]: False or True. f True, it will expand double the number of solutions that the program would normally expand.
Execution
Interactively:
ARCIMBOLDO_SHREDDER input.bor
In background:
When using a grid in which a password is required, it may be given as input in a text file (e.g. password). Otherwhise, just redirecting the output and using & will launch the job in background.ARCIMBOLDO_SHREDDER input.bor < password >& log &
Output
Sequential mode
For each rotation cluster, shredder will generate five ARCIMBOLDO search models. The ARCIMBOLDO-SHREDDER work folder contains a set of folders called ARCI_*/, where * refers to the number of the rotation cluster. Each contains five folders with ARCIMBOLDO runs called after the search model used. Models are saved into the ./library/ folder. You will find:
- peaks: a run using the peaks_*_0.pdb model, that is, the model created by selecting the peaks determined in the Shred LLG function.
- overt: a run using the overt_*_0.pdb model, that is, the model created by discarding all residues that are above the minimum peak height in the Shred LLG function.
- percentile70: a run using the percentile70_*_0.pdb model, that is, the model created by discarding all residues that are above the 70th percentile in the Shred LLG function.
- percentile75: a run using the percentile75_*_0.pdb model, that is, the model created by discarding all residues that are above the 75th percentile in the Shred LLG function.
- pklat: a run using the pklat_*_0.pdb model, that is, the model created eliminating both the peaks selection and plateau regions.
Each of the folders will contain an html file with the ARCIMBOLDO run output.
Spherical mode
All generated models are evaluated as a library in an ARCIMBOLDO_BORGES run. In the working directory there is a folder called ARCIMBOLDO_BORGES and one called models. Models contains the library, and ARCIMBOLDO_BORGES is a run of ARCIMBOLDO_BORGES using that library. In the working directory there is an html file, but only contains the echo of the bor file and a link to the ARCIMBOLDO_BORGES run html. Inside the ARCIMBOLDO_BORGES folder is where the best* files and the html of that run will be found.
Go to top page
ALEPH manual
Execution:
The ALEPH software can be run from its Graphical User Interface or from the command line. It requires Python3.
Graphical User Interface:
ALEPHUI
Command line:
The generic command line will be composed by the following elements:ALEPH ALEPH_mode --parameter1 parameter1_value --parameter2 paramter2_value...
Input:
ALEPH_mode: annotate (A), decompose (D), generate_library (L) or superpose (S).
Note: there is another ALEPH_mode parameter called find_folds. Activation for a special mode of decompose in which is perform a iterative hierarchical clustering.
Parameters are dependent of the mode selected. The following list contains all the parameters and their description. Mode(s) where they apply are specified in parentheses. Default values and parameter range are quoted:
[pdbmodel](A,D,L): /path/to/model_file.pdb. Pdb model for input.
[strictness_ah](A,D,L,S): 0.5; 0-1. Strictness threshold for accepting ah CVs. High values are used for a precise annotation while selection of low values extend secondary structure elements.
[strictness_bs](A,D,L,S): 0.3; 0-1. Strictness threshold for accepting bs CVs. High values are used for a precise annotation while selection of low values extend secondary structure elements.
[algorithm](D): fastgreedy, infomap, eigenvectors, label_propagation, community_multilevel, edge_betweenness, spinglass, walktrap. Algorithm for the community clustering procedure.
[homogeinity](D): False or True. If True, homogeneously sized clusters are favoured.
[pack_beta_sheet](D): False or True. If True, avoids splitting a beta sheet in different groups.
[work_directory](L): /path/to/working/directory.
The dataset from which generate the library can be restricted in different ways:
[directory_database](L): Set the database as the given input folder.
[cath_id](L): Extract from the PDB a database of the given cath id.
[target_sequence](L): /path/to/sequence_file.seq. Extract a database from the given target sequence. The sequence is used to perform a BLAST search and for significant hits the SCOP_ID and CATH_ID are read, then structures of the same CATH_ID or SCOP_ID are downloaded to generate the input database.
ALEPH generation of libraries can be parallelized. Default running is in multiprocessing but the following options are available:
[supercomputer](L) /path/to/configuration_file.bor.
[remote_grid](L) /path/to/configuration_file.bor.
[local_grid](L) /path/to/configuration_file.bor.
[score_intra_fragment](L,S): 95; 0-100. Global geometrical secondary structure match expressed as score percentage.
[score_inter_fragments](L,S): 90; 0-100. Global geometrical tertiary structure match expressed as score percentage.
[rmsd_min](L): 0.0. Superposition threshold, minimum rmsd against the template.
[rmsd_max](L): 6.0. Superposition threshold, maximum rmsd against the template.
[clustering_model](L): no_clustering, rmsd, rmsd_range, random_sampling. Clustering modes in library generation.
[rmsd_clustering](L, rmsd): Threshold for pairwise comparison.
[number_of_ranges](L, rmsd_range): 500. Number of groups.
[number_of_clusters](L, rmsd_range, random_sampling): 7000. Number of representative models extracted from the library.
[exclude_sequence](L): False or True. If True, avoid the extraction of models from any chain that aligns within a s.i. >= 90% from this sequence.
[test](L): False or True. If True, test with a reduced sample of models to check parameterisation.
[representative](L): False or True. If True, for each structure in the PDB database extracts only the model with lowest rmsd.
[reference](S): /path/to/model_file.pdb. Reference pdb model fixed in superposition.
[target] /path/to/model_file.pdb or [targets] /path/to/directory (S): Target pdb model or directory of pdb models for moving structure in superposition.
[rmsd_thresh](S): 1.5. Rmsd threshold to accept a superposition.
[peptide_length](A,D,L,S): 3. Peptide length for computing a CV.
[width_pic](A,D,L,S): 100.0. Width in inches for pictures.
[height_pic](A,D,L,S): 20.0. Height in inches for pictures.
[min_ah_dist](A,D,L,S): 0.0. Minimum distance allowed among ah CVs in the graph.
[max_ah_dist](A,D,L,S): 20.0. Maximum distance allowed among ah CVs in the graph.
[min_bs_dist](A,D,L,S): 00.0. Minimum distance allowed among bs CVs in the graph.
[max_bs_dist](A,D,L,S): 15.0. Maximum distance allowed among bs CVs in the graph.
[write_graphml](A,D,L,S): False or True. If True, write graphml files.
Output:
ALEPH outputs annotated secondary structure fragments in a pdb file and plots geometrical properties of CVs in png files. The decomposition mode marks the groups with different chain IDs in the pdb file. If library generation is performed, ALEPH outputs the pdb files of the superposed models in a new directory called library. In cases when clustering is activated, another directory called clusters is written containing the representative models. The superposition outputs a pdb with the superposed target. All these files are displayed through ALEPHUI.
Go to top page
ALIXE manual
Overview:
A phase combination tool to increase the signal of partial solutions while reducing redundancy, to promote structure solution in difficult cases. Of particular interest for fragment-based methods such as the ARCIMBOLDO programs.
Usage:
ALIXE can be run directly through the ARCIMBOLDO programs setting the keyword alixe to True in the .bor configuration file of the ARCIMBOLDO run. In ARCIMBOLDO-SHREDDER it is activated by default in monomer mode.
Alternatively, ALIXE can be called from the command line:
-
ALIXE.py -m mode -i input_folder or arcimboldo_bor -c path_to_chescat_executable -d hkl_file -s pdb_symmetry (-d and -s are optional keywords depending on data in input_folder).
-
ALIXE.py name.bor
The use of a configuration bor file allows the management of the whole parameterization given to ALIXE.
Interactively:
ALIXE input_alixe_bor
In background:
Redirecting the output and using & will launch the job in the background and produce a log file with the screen output.ALIXE input_alixe_bor >& log &
The following line generates a template for an annotated ALIXE configuration file: ALIXE.py -f name_conf.bor.
Mandatory input
ALIXE accepts phase information from maps in SHELX .phs format, or coordinate files (.pdb or .pda) of the partial solutions. In the first option ALIXE will require:
- Maps in SHELXE .phs format derived from partial solutions, ideally after 5 cycles of density modification to enhance recognition of the origin shift.
- A pdb or pda file with a CRYST1 card to retrieve the unit cell and space group.
- Path to the CHESCAT executable.
For the second option ALIXE will require:
- Coordinates in pdb or pda format corresponding to the partial solutions.
- A SHELX reflection file (hkl).
- Path to the CHESCAT executable (if not set).
- Path to SHELXE (if not set).
Input can be given to ALIXE in two ways:
- By command line using the mandatory keywords:
- -m mode of ALIXE
- -i followed by either a /path/to/input_folder or /path/to/arcimboldo_bor_file
- -c /path/to/the_CHESCAT_executable
- -d /path/to/file.hkl (SHELX reflection file)
- -s /path/to/file.pda or /path/to/file.pdb with a CRYST1 card if phase sets are provided as .phs files
- Using a configuration file for ALIXE (.bor), where not only the mandatory input but also all other parameters can be modified.
Remaining parameters will be set to default values when using command line. Additional optional arguments are:
The following parameters are mandatory and should be input by the user:
[input_info_1]: /path/to/input_folder or /path/to/arcimboldo_bor_file.
[input_info_n]: n as the number of folders given.
[output_folder]: a name which should be unique to the run and contain no spaces.
[alixe_mode]: monomer, multimer, cc_analysis, fish
If fish mode selected:
[references]: none or /path/to/reference_solutions. Use this solution or solutions as references only.
Path to the local executables:
[path_chescat]: /path/to/chescat
[shelxe_path]: /path/to/shelxe
The following is only required if solutions are given in pda or pdb format and phs must be computed:
[hkl_file]: /path/to/file.hkl (data in SHELX hkl format)
The following is only required if solutions are given in a folder in a SHELX .phs format:
[path_sym]: /path/to/file.pda or /path/to/file.pdb (file providing the symmetry information)
The following parameters can be modified but are not mandatory because they have preset defaults (bold). They are presented in two groups (basic and advanced):
Basic
[tolerance_first_round]: 60. wMPD value in degrees used as a threshold to combine phase sets. If wMPD between phase sets is below that value they will be combined.
[tolerance_second_round]: 87. wMPD value in degrees used as a threshold to combine the clusters produced in a first round. Only in the multimer mode.
[expansions]: False or True. If set to True, expansions will be sequentially launched.
[plots]: True. Graphical output will be computed.
Advanced
[resolution_comparison]: 4.0. Resolution in Å at which the phase sets will be compared. If the crystal belongs to the space group P1 is recommended to use 3.5 Å.
[resolution_merging]: 2.0. Resolution in Å at which the phase sets that were found similar at the comparison are merged to produce the cluster that will be used for density modification and autotracing.
[seed]: 1 or 0. It refers to the sequential part of the algorithm. 1 reduces intercalls between chescat and ALIXE when processing the list of phase sets speeding up the process.
[origin_search]: sxosfft or sxos. Algorithms for the determination of the origin shift.
[weight]: e or f. Weight to apply to the reflections. It can be E (normalized amplitudes) or F (amplitudes).
[oricheck]: True or False. If False, the origin shift will not be checked after cycle 1.
[cycles]: 3. Number of cycles of iterative clustering.
[minchunk]: 100. Number of remaining phase sets to shift from the parallel part of the algorithm to the sequential part.
[max_non_clust_events]: 20. Number of iterations producing no cluster after which the parallel algorithm shifts to the sequential.
[fom_sorting]: COMBINED, CC, LLG or Z-score. Figure of merit used to sort the phase sets prior to the clustering.
[fragment]: 1. Integer in the range of the number of fragments, indicating from which fragment should the solutions for ALIXE be extracted Only for ARCIMBOLDO_LITE runs.
[map_cc]: True or False. If True, map correlation coefficient will be computed.
[number_cores_parallel]: -1. Number of cores to exploit in parallel parts of the algorithm. If -1 it will exploit all the number of logical cores available in the workstation minus one up to 10 cores.
[fusedcoord]: False or True. If True, generates in real space the equivalent merging of solutions.
[ccfromphi]: False or True. If True, computes the initial CC of the combined phases with SHELXE.
[postmortem]: False or True. If True, a reference file in pdb format must be provided with the extension .ent and ALIXE will perform a wMPE assessment of the solution
Output
ALIXE outputs a log file:
Command line used for autoalixe, started at 1582895117.76 was /ARCIMBOLDO_FULL/ALIXE.py conf_alixe.bor Total time spent in running autoalixe is 140.993822098 seconds, or 2.34989703496 minutes Command line used was /ARCIMBOLDO_FULL/ALIXE.py conf_alixe.bor
It echoes the command line used and gives time information.
There is also a folder called clustpool_n containing the clusters produced with the .phi extension named after the reference used to form it. Information about the phase sets joined in each cluster appears in the summary text files (.sum).
| Name | wMPD_first | wMPD_last | diff_wMPD | mapcc_first | mapcc_last | shift_first_x | shift_first_y | shift_first_z | shift_last_x | shift_last_y | shift_last_z |
|---|---|---|---|---|---|---|---|---|---|---|---|
| frag548_0_0_rbr_11.phs | 51.5 | 35.6 | -1 | 43.56 | 61.17 | 0.0 | 0.0 | 24 | 0.0 | 0.0 | 0.0 |
| frag556nogyre_0_0_rbr_11.phs | 48.7 | 25.8 | -1 | 45.64 | 72.81 | 0.0 | 0.0 | -491 | 0.0 | 0.0 | -0.0 |
| frag508nogyre_0_0_rbr_11.phs | 52.1 | 21.9 | -1 | 42.81 | 78.3 | 0.0 | 0.0 | -0.48 | 0.0 | 0.0 | 0.0 |
| frag598_0_0_rbr_11.phs | 50.2 | 44.9 | -1 | 44.33 | 50.34 | 0.0 | 0.0 | 69 | 0.0 | 0.0 | -0.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
Table 1. Summary file.
The summary file above contains the phase sets combined in one particular cluster, the MPD and the map correlation coefficient between the phase sets in the first cycle of search of the origin shift and in the final one. It also shows the origin shifts applied to the phase sets.
The higher the similarity between the phase sets, the lower their wMPD. Only phase sets with an wMPD below the threshold set will be combined in a cluster.
If the keyword fusecoord is set to True it will generate in real space the equivalent merging of solutions in coordinate ending with the termination '_shifted.pda'. In those files, the partial solutions have been shifted using the values found in their phase comparison.
ALIXE outputs two instructive tables gathering the most relevant information available about your partial solutions and clusters: clustpool_n_info_frag and clustpool_n_info_clust_table.
| Name | LLG | Z-score | Rotcluster | InitCC | Efom | PseudoCC |
|---|---|---|---|---|---|---|
| frag461_0_0_rbr_3 | 27.5 | 4.7 | 3 | 6.01 | 0.181 | 21.28 |
| frag288_0_0_rbr_11 | 35.9 | 4.25 | 11 | 6.09 | 0.211 | 24.91 |
| frag171nogyre_0_0_rbr_11 | 22.0 | 4.34 | 11 | 5.38 | 0.182 | 21.88 |
| ... | ... | ... | ... | ... | ... | ... |
Table 2. Clustpool_n_info_frag.
| Cluster | n_phs | topzscore | topllg |
|---|---|---|---|
| frag588_0_0_rbr_11_ref.phi | 20 | 8.17 | 71.70 |
| frag330nogyre_0_0_rbr_11_ref.phi | 2 | 4.89 | 25.20 |
| frag231nogyre_0_0_rbr_3_ref.phi | 2 | 4.61 | 25.20 |
| frag218nogyre_0_0_rbr_3_ref.phi | 2 | 4.29 | 23.40 |
| frag9_0_0_rbr_3.phs | 1 | 4.36 | 40.10 |
| frag79nogyre_0_0_rbr_0.phs | 1 | 4.18 | 25.80 |
| ... | ... | ... | ... |
Table 3. Clustpool_n_info_clust_table.
These tables display figures of merit for single solutions and for the phase clusters, respectively.
In addition, it plots several graphs to procure a general idea of the landscape of solutions used considering their scores if available. Thus, it plots clustpool_1_single_sol_Z-score_LLG.pdf and clustpool_1_single_sol_InitCC_LLG.pdf representing all single solutions and clustpool_1_info_allclust_LLG_vs_Zscore.pdf and clustpool_1_info_allclust_LLG_vs_FusedCC.pdf representing the single solutions and the clusters composed.
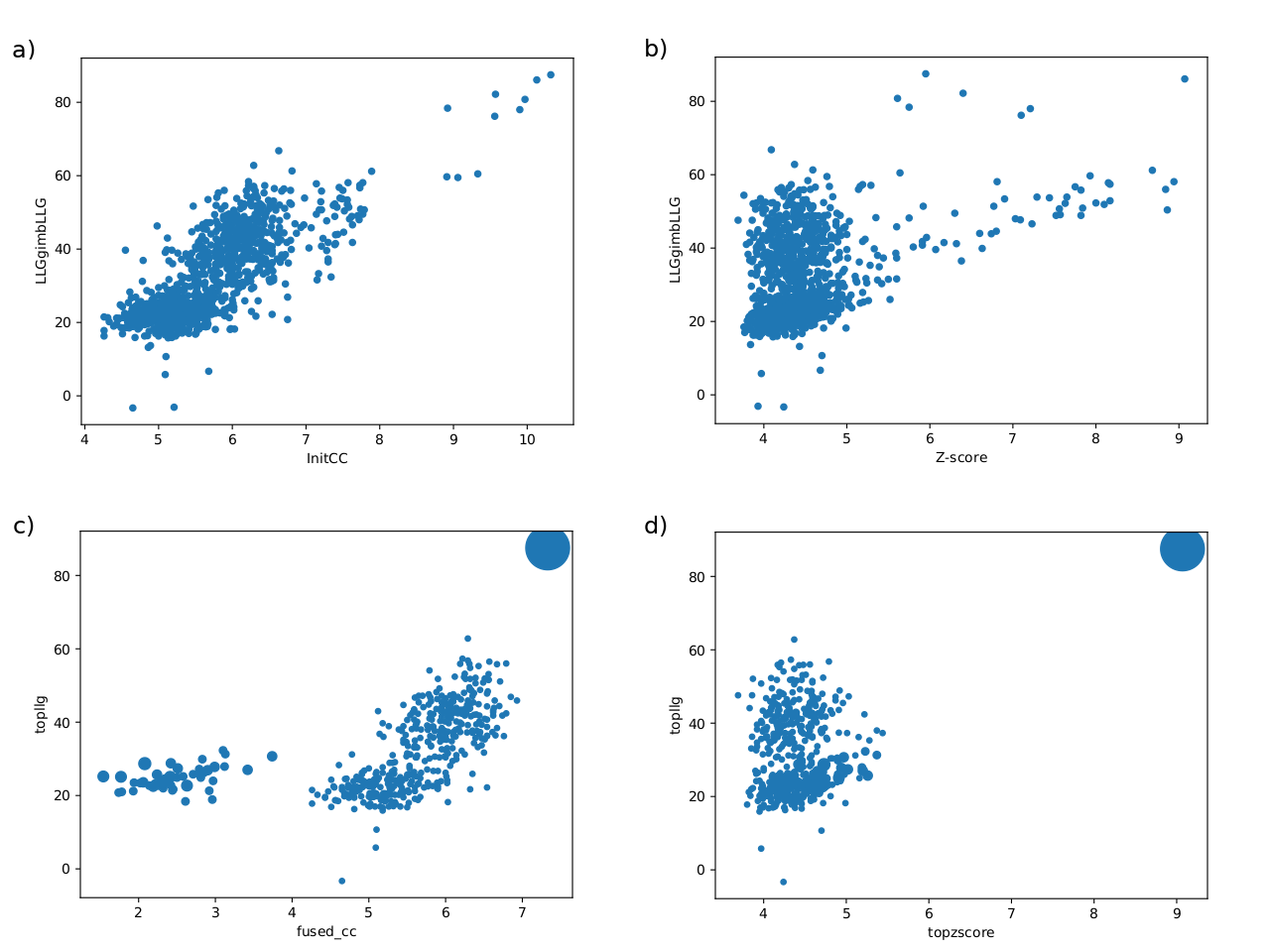
Figure 1.
Examples illustrating typical landscapes of partial solutions rendered by phasing with fragments. The points in the scatter plots represent either single partial solutions (a)(b) or clusters of partial solutions (c)(d) from an ARCIMBOLDO-SHREDDER run. The ordinate shows the LLG score after rigid-body refinement and the abscissa shows the correlation coefficient (a)(c) and the Z-score (b)(d).
(a)(b) Plots representing the distribution of single partial solutions.
(c)(d) Plots representing the distribution of clusters.
In these particular plots, the single, large cluster produced, showing top figures of merit conforms a typical positive outcome.
These plots provide a global landscape of the partial solutions, suggesting how probable are correct solutions within the whole set. A promising distribution will show a clear distinction among probes, with single partial solutions (Figure 1(a), 1(b)), characterized by better figures of merit, clearly detached from the bulk of wrong probes.
They are complemented by plots representing the resulting phase clusters. These illustrate the reduction of partial solutions. The radius of the circles is proportional to the number of phase sets that have been combined in each resulting cluster. Larger circles (joining many phase sets) might be indicative of success because true phase sets should be consistent with each other when referred to a common origin. Additionally, the clusters are plotted reflecting the top score reached by one of their phase sets. Large clusters, showing clear discrimination provide a promising indication and if the keyword expansions is set to True, these clusters will be trialled for density modification and autotracing, possibly leading to a correct and complete solution. Nevertheless, as in fragment-based molecular replacement probes are not independent in their generation, indications might be misleading.
Go to top page
SEQUENCE SLIDER manual
Overview
An ARCIMBOLDO or Molecular Replacement partial solution with promising Figures of Merit are particularly suited for SEQUENCE SLIDER. If partial solution comes from a remote homologous (such as coming from ARCIMBOLDO_SHREDDER), the alignment should be given to restrict hypotheses generation by SLIDER (Remote-Homolog Mode), otherwise secondary structure prediction from PSIPRED should be given (Secondary-Structure Mode).
Input
- An mtz file containing the reflection data.
- A SHELX reflection file hkl containing the reflection data.
- A pdb with partial solution.
- The configuration .bor file with the parameters for an SEQUENCE SLIDER run, which is defined as follows:
[CONNECTION]: #Values for the following keyword are mutually excluding distribute_computing: multiprocessing #distribute_computing: local_grid #distribute_computing: remote_grid #default is to search your rsa private keyfile into ~/.ssh/id_rsa #remote_frontend_passkey: #setup_bor_path: [GENERAL]: working_directory: mtz_path: hkl_path: pdb_path: [SLIDER] #if run is in Secondary Structure Mode, then secstr_path is compulsory, #else Remote Homologue Mode requires align_path secstr_path: align_path: molecular_weight: number_of_component: #i and sigi_label inclusion are recommended i_label: sigi_label: #f and sigf_label are compulsory if refinement_program buster or refmac are used f_label: sigf_label: rfree_label: refinement_program: # should be buster / phenix.refine / refmac #The following section is only required in multiprocessing mode [LOCAL] path_local_phaser: path_local_shelxe: path_local_sprout: # if refinement_program: buster path_local_buster #path_config_buster # if refinement_program: phenix.refine path_local_phenix.refine: #path_config_phenix: # if refinement_program: refmac path_local_refmac: #path_config_ccp4
Optional keywords:
[LOCAL] path_local_edstats: [SLIDER]: name_job: chosen_chains: ncschains: False trust_loop: False RandomModels: 0 RenumberResidues: True psipred_confidence_level: 0 psipred_min_frag_size: 4 minimum_ss_frag: 5 sliding_tolerance: 3 models_by_chain: 100 #maximum 3000 seq_pushed_refinement: 100 #maximum 1000 ModelEdge: False ReduceComplexity: False Recover0occup: False fixed_residues_modelled: shelxe_line: number_shelxe_trials: 15 expand_from_map: False refmac_parameters_path: buster_parameters: -noWAT -nbig 10 -RB -nthread 1 UsePdbchk="no" PhenixRefineParameters: strategy=individual_sites+individual_adp+individual_sites_real_space+rigid_body main.number_of_macro_cycles=5 write_eff_file=false write_geo_file=false write_def_file=false optimize_xyz_weight=False optimize_adp_weight=False nproc=1 export_final_f_model=true sspdb_path: #should be a file containing chain : and SS assignment (H for helix, E for strand and C for coil), example: "A: HHHH"
[CONNECTION]
[distribute_computing]: multiprocessing or local_grid or remote_grid. Default is multiprocessing on a single machine. If a grid is used, the next two variables should be defined.
[setup_bor_path]: path to the configuration file for program setup.
[remote_frontend_passkey]: False. If you want to use your personal id_rsa key that is not stored in the default path ~/.ssh/id_rsa, then put the full path here.
[GENERAL]
[mtz_path]: path to the mtz file with reflection data.
[hkl_path]: path to the hkl file with reflection data.
[pdb_path]: path to the pdb file with partial solution.
[ent_path]: path to the ent file with solved structure, should be in the same asymmetric unit.
[working_directory]: absolute path to working directory.
[SLIDER]
[secstr_path]: path to the PSIPRED output, compulsory in Secondary Structure Mode.
[align_path]: path to the alignment, compulsory in Remote Homologue Mode.
[molecular_weight]: it will be used by PHASER to calculate the composition in the ASU, assuming that protein nucleic acid have the average distribution of amino acids and bases.
[number_of_component]: number of copies of protein/nucleic acid defined by the molecular weight.
Latest Phaser versions are able to work directly with intensities, because new likelihood targets and functions have been defined. We strongly recommend to use this feature if possible. For that purpose, use the keywords i_label and sigi_label in the bor file to indicate the columns from the mtz.
[i_label]: label for the intensities in the mtz file.
[sigi_label]: label for the standard deviation of the intensities in the mtz file.
[f_label]: label for the amplitudes in the mtz file.
[sigf_label]: label for the standard deviation of the amplitudes in the mtz file.
[LOCAL]
[path_local_phaser]: path to your local PHASER installation.
[path_local_shelxe]: path to your local SHELXE installation.
[path_local_sprout]: path to your local SPROUT installation.
[path_local_buster]: path to your local BUSTER installation.
[path_config_buster]: path to your local BUSTER source file.
[path_local_refmac]: path to your local REFMAC5 installation.
[path_config_ccp4]: path to your local CCP4 source file.
[path_local_phenix.refine]: path to your local PHENIX.REFINE installation.
[path_config_phenix]: path to your local PHENIX source file.
Advanced and optional keywords
[LOCAL]
[path_local_edstats]: path to your local EDSTATS installation.
[SLIDER]
[name_job]: string, should be unique to the run and have a max length of 20 non-special characters except "_" (underscore).
[chosen_chains]: chains whose side chains atoms will be assigned. If assignment will be simultaneously to more than one chain, they should be separated by comma (,). Otherwise, independent chains should be separated by dot comma (;).
[ncschains]: if True, NCS-related chains should be given in [chosen_chains] separated by commas (,).
[trust_loop]: if True, loop residues will also receive side chain assignments.
[RandomModels]: number of models with random sequence to be included in run.
[RenumberResidues]: if True, number of residues will be changed in accordance with sequence hypothesis, otherwise numbering will be the same for all models and identical to initial model.
[psipred_confidence_level]: confidence level requirement for considering alpha-helix or beta-strand in PSIPRED.
[psipred_min_frag_size]: minimum size of considered secondary structure fragment from PSIPRED.
[minimum_ss_frag]: minimum size of fragment in [pdb_path] to be included in side chain assignment.
[sliding_tolerance]: how many slides SEQUENCE WINDOW will perform. If Remote Homologue Mode and value 1 is given, it will generate three sequences from alignment, its exact correspondence, 1 residue slide to the left and 1 to the right. If Secondary Structure Mode and 1 is given, then fragments diverging in size of 1 will be included in the assignment.
[models_by_chain]: number of hypotheses to be pushed to side chain modeling.
[seq_pushed_refinement]: number of hypotheses to be pushed to refinement.
[ModelEdge]: if False, edges of fragment in [pdb_path] and in [chosen_chains] will have an Ala assignment.
[ReduceComplexity]: if True, only hydrophobic residues will be assigned to hypotheses (i.e., AFGILMPVW).
[Recover0occup]: if True, residues possessing 0 occupancy in [pdb_path] will be kept.
[fixed_residues_modelled]: chain and residue numbers of amino acids that should be retrieved from the initial model and kept in model, otherwise Ala will be used. It should be given as chain:residueN1-residueN2,residueN3-residueN4 such as: A:1-10,20-30 B:1-5,30-35 ; residues in this range will be modelled.
[shelxe_line]: SHELXE parameterization for autotracing.
[expand_from_map]: if True, maps from refinement will be input in SHELXE for autotracing.
[refmac_parameters_path]: path of file containing instructions for REFMAC refinement.
[buster_parameters]: parameters for BUSTER refinement.
[PhenixRefineParameters]: parameters for PHENIX.REFINE refinement.
[sspdb_path]: if secondary structure assignment of ALEPH in [pdb_path] and [chosen_chains] is not desired, this information may be overwritten following H for helix, E for strand and C for coil, example of a 4-residue alpha-helix: "A: HHHH".
Execution
Interactively:
SLIDER.py input.bor
In background:
When using a grid in which a password is required, it may be given as input in a text file (e.g. password). Otherwise, just redirecting the output and using and will launch the job in background.SLIDER.py input.bor < password >& log &
Output
The .html output file created will summarize the one of the most relevant figures of merits (FOM) from SEQUENCE SLIDER, the log-likelihood gain (LLG) calculated by PHASER and the correlation coefficient (CC) from SHELXE. If the expand_from_map option is set to True, expansions from coordinates are numbered 0 and shown in 2nd plot and from maps, 1, shown in third plot. The FOMs of the initial model (star in graph) gives a baseline in respect to the models receiving side chains (triangles in graph). If random sequences (diamonds in graph) are assigned to the initial model, their FOM gives a base line of true negative, even though there will be some correctness in main chain atoms and some overlap in side chain atoms.
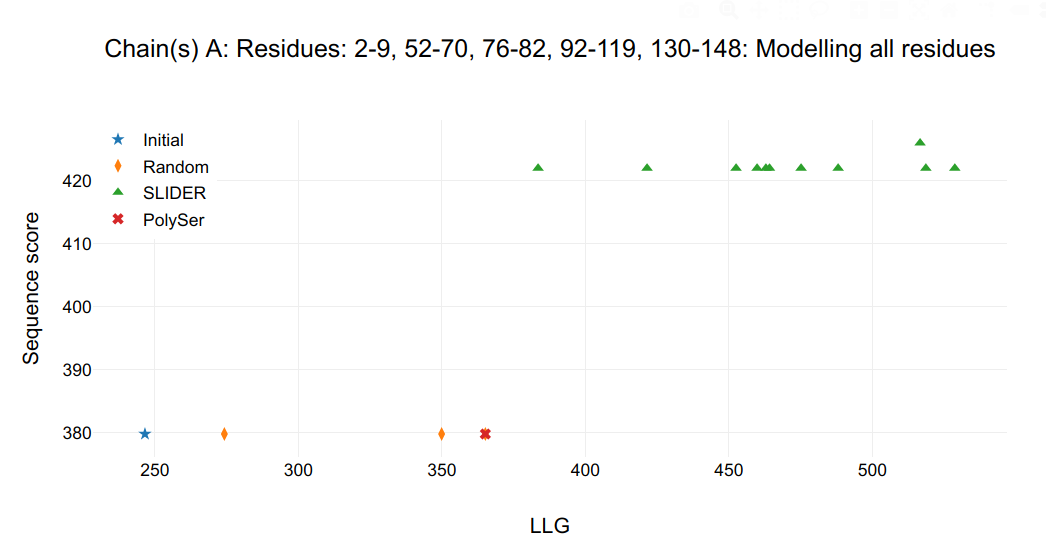
Figure 1. Refinement statistics.
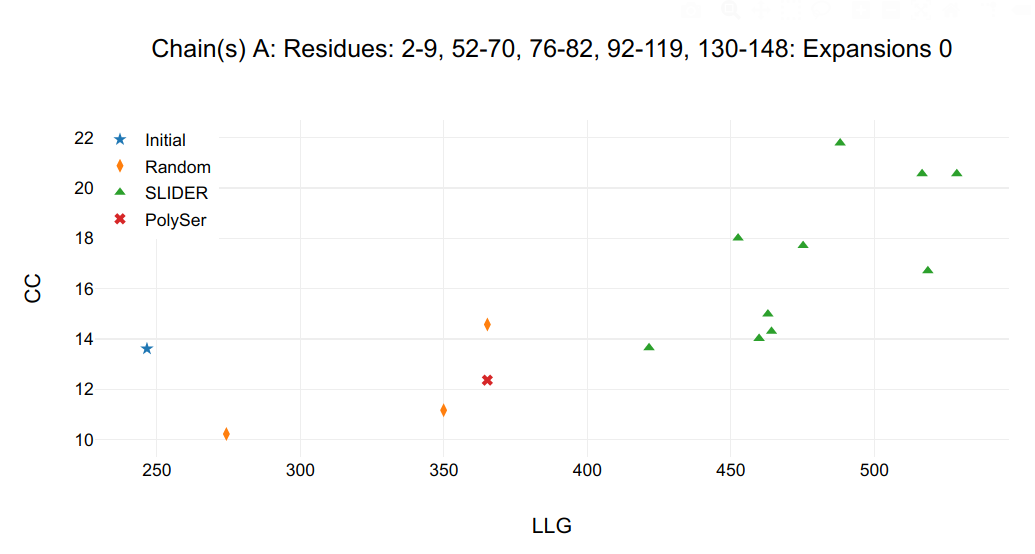
Figure 2. Expansion statistics.
Additional FOM from model refinement, such as Rfactors (R and Rfree) and real-space correlation coefficient of main chain atoms (RSCCmc) and RSCC of side chain atoms (RSCCsc) may be found at the output directory of SLIDER (working_directory/SLIDER/seq*/Table_refine1_seq*.log). Also this log file contains the number of residues having side chains assigned (assigne), PHASER LLG (LLG), and delta contrast of LLG (DContr). DContr the number of standard deviations from the mean LLG obtained for all models involving the same chain(s). If phenix.refine was chosen in refinement_program option, LLGnorm, LLGwork and LLGfree from refinement are also shown in this log file.
Additional FOM from expansion, such as final number of residues (nres), number of traced chains (nchains) and CC from trace may be found at the output directory of SLIDER (working_directory/SLIDER/seq*/Table_expansion_seq_*.log ). If expand_from_map option is set to True, expansions from coordinates are numbered 0 and from maps, 1.
Empirically, depending on the data and model completion, a model scoring a high LLG in comparison to rest can be enough to distinguish correct sequence assignment. From the tests shown in the main citation of SEQUENCE SLIDER, we have seen that if its 1σ contrast is 1σ above the rest of models, it is sufficient to distinguish the correct hypothesis. This is particularly interesting for structures whose sequence is difficult to assign, such as coiled coils.
Go to top page