ARCIMBOLDO for MicroED tutorial
Aims of the tutorial: Using ARCIMBOLDO to solve MicroED data
The aim of the following tutorial is to exemplify the use of ARCIMBOLDO_SHREDDER on data from the micro electron diffraction technique. Both SHREDDER sequential and SHREDDER spheres will be using the dataset deposited under the PDB id 6V8R, corresponding to 1.6 Å microED data from the proteinase K.
Preliminary considerations relevant to phasing MicroED data using the ARCIMBOLDO programs:
- Phaser supports the usage of tabulated electron scattering form factors, but it is not the default, and therefore the configuration file of the three ARCIMBOLDO programs (LITE, BORGES and SHREDDER) has a new keyword, called formfactors, that must be set to FORMFACTORS ELECTRON, for ARCIMBOLDO to call Phaser with the appropriate setting.
- SHELXE: One of the most powerful underlying assumptions in density modification is that the density determined by X-ray diffraction should never be negative, so all negative density in the macromolecule region is set to zero. However for electron diffraction the electrostatic potential map is partly positive and partly negative (from the negative charges), so density modification would be less effective.
- Data correction is critical, anisotropy correction should filter reflections on information content1,2 or apply an anisotropic resolution cutoff. In either case, the flags -e and -I should be used in SHELXE to make sure missing reflection will be extrapolated in every cycle, to avoid deterioration of the maps.
Experimental details and data
Test data for our tutorial have been obtained from the proteinase K (PDB ID: 6V8R), collected and processed by José Rodríguez’s group in UCLA3. The asymmetric unit contains one monomer of 279 residues.
Characteristic of the dataset extending to 1.6 Å are summarized in the following table:
| Data collection and Processing | |
|---|---|
| Resolution (Å) | 55.79-1.6 (1.657-1.6) |
| # crystals | 6 |
| Electron Dose (electrons per Å**-2) | 0.0357 |
| Molecular Weight (kDa) | 28.9 |
| Space Group | P43212 |
| a, b, c (Å) | 67.25, 67.25, 99.92 |
| A, B, and C (degrees) | 90,90,90 |
| # total reflections | 194052 |
| # unique reflections | 29,058 (2,506) |
| CC1/2 | 0.912 (0.051) |
| <I/σI> | 3.31 |
| Completeness | 91.49 (66.19) |
| Multiplicity | 6.68 |
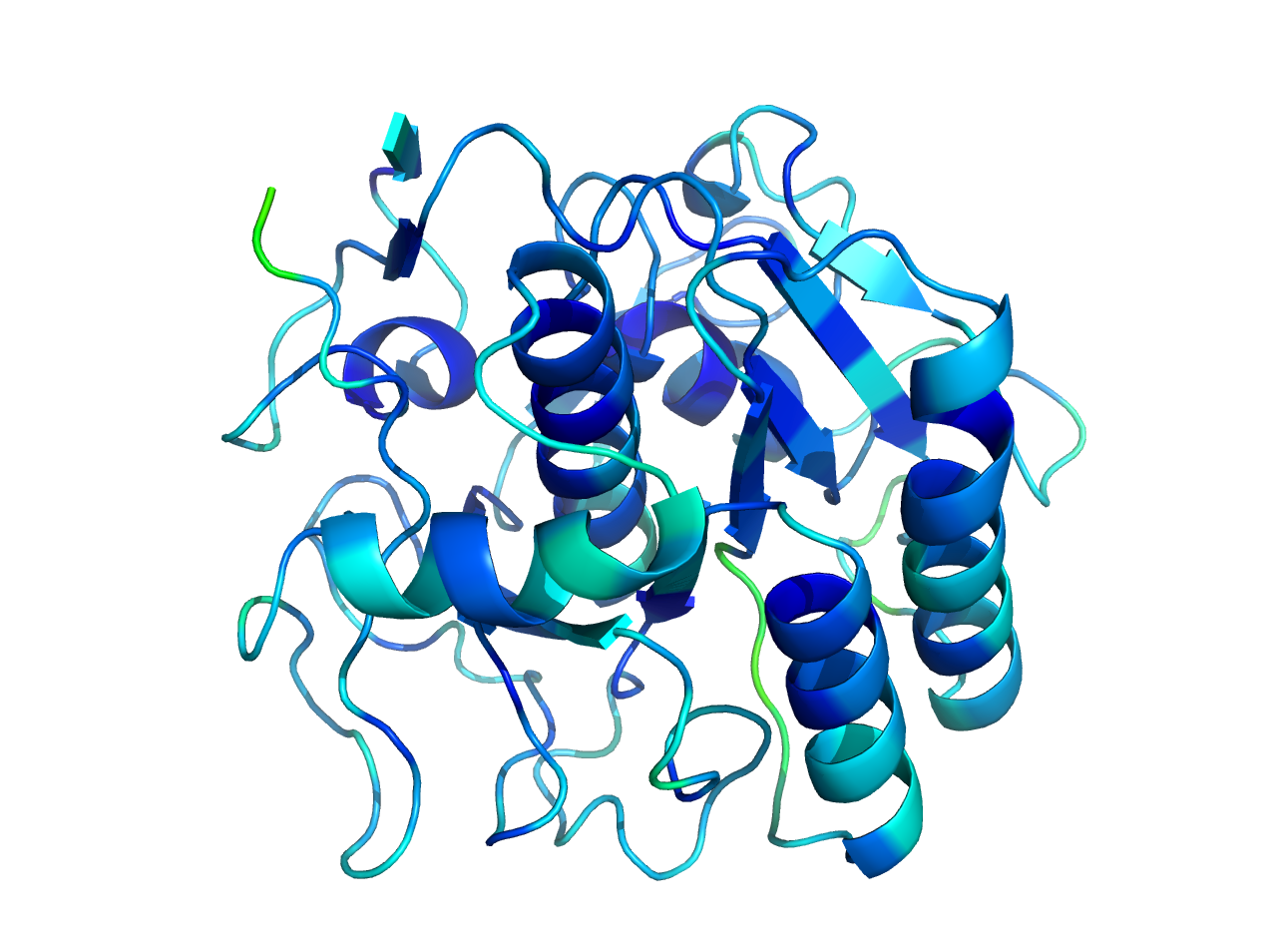
Figure 1. Structure of Proteinase K colored after B-value.
Solving proteinase K with the sequential mode in ARCIMBOLDO_SHREDDER
Step by Step tutorial
1. Creation of the library from a selected homology model
The model chosen was the Aqualysin I from Thermus aquaticus YT-1 (deposited under the PDB id 4DZT) and with a 40% sequence identity to the target structure.
Input
We will need:
All required files can be downloaded here.
The description of the configuration file follows:
[CONNECTION]: distribute_computing: multiprocessing [LOCAL] path_local_phaser: /path/to/local_phaser path_local_shelxe: /path/to/local_shelxe [GENERAL]: working_directory: /path/to/working_directory mtz_path: %(working_directory)s/6v8r.mtz hkl_path: %(working_directory)s/6v8r.hkl [ARCIMBOLDO-SHREDDER] name_job: 6v8r molecular_weight: 28900 i_label: IMEAN sigi_label: SIGIMEAN formfactors: FORMFACTORS ELECTRON trim_to_polyala = True rmsd_shredder: 1.2 model_file: /absolute/path/to/4dzt.pdb SHRED_LLG: True SHRED_METHOD: sequential SHRED_RANGE: 10 20 4 fragment shelxe_line = -m15 -a8 -s0.5 -v0 -t10 -q -o -y1.60 shelxe_line_last = -m15 -a1 -s0.45 -v0 -t10 -q -o -y1.60 -e1.10
In the .bor file you need to specify:
In the [ARCIMBOLDO-SHREDDER] section you will need to define the contents of the asymmetric unit, the model pdb to use, the number of copies to search and the labels for the mtz file.
The keywords SHRED_LLG, SHRED_METHOD and SHRED_RANGE: 10 20 4 fragment constitute the key part of this section, defining the shredding, in this case a sequential shred from 10 to 20 residues with step size of 4. These are not default values but produce one fourth of the possible fragments, making the computation faster. As we input the keyword fragment, models will be generated extracting the shreds of 10,14, etc residues at every possible starting position. If we had used the keyword “omit” instead, resulting fragments after omitting shreds of 10, 14,.. residues would be probed.
The expected similarity between target and model is expressed for its use in Phaser through two rmsd values. One is used in the Shred-LLG evaluation and optimization and a second one for the ARCIMBOLDO runs launched with the models generated in the first step. Considering that models should improve with respect to the original template, it is advisable to use a smaller value for the rms deviation for ARCIMBOLDOs than for SHREDDER. If no values are provided, default values will be used. In this case the program will use the specified value for SHREDDER and the default value of 0.8 Å for the subsequent ARCIMBOLDO_LITE searches with the models derived from SHREDDER, as it is not specified in the bor file.
Execution
You can run the program interactively or in the background, redirecting the output to a log file.
1. Interactively
ARCIMBOLDO_SHREDDER 6v8r.bor
2. In background
nohup ARCIMBOLDO_SHREDDER 6v8r.bor >& log &
Output and Results
In the directory where you launched ARCIMBOLDO_SHREDDER, you have a ./library/ folder containing all models and a set of folders called ARCI_*/, where * refers to the number of the rotation cluster. For this case, only a single rotation cluster (cluster 0) in the preliminary evaluation of the whole template is discriminated enough to be selected. Its models are used for running the posterior ARCIMBOLDO_LITE searches. Inside the ARCI_0 folder, there is another set of sub-folders, called overt, peaks, percentile70, percentile75 and pklat. You will find inside of each of them an html file summarising the ARCIMBOLDO run.

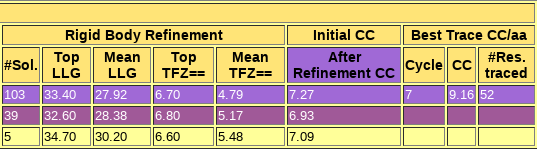


Figure 2. a) Output from the peaks model b) Output from the overt model. This figure shows the contrast between a run with a clear solution (b), where the figures of merit are really high and a single solution is found, and an unsuccessful run with multiple rotation clusters and multiple solutions that do not discriminate.
You can check the html output here. The first section echoes all parameters used for the run, so that defaults are listed along with those set in the .bor file. This allows reproducing the run even if defaults may change in future versions. The next section displays a sortable table summarizing the results for each step. The percentile70, percentile75 and overt models produce clear, single solutions with high figures of merit as it can be appreciated in the tables in the html. Moreover, their wMPE to the true phases of 6V8R is between 60.4º and 62.8º. The html file also lists the backtracking for the best solution (in this case, a single solution). Automatic interpretation of the correctly placed fragments in terms of density modification and autotracing with SHELXE is challenging for the issues described before, and the complete building and refinement of the structure starting from this best solution involves a few more steps that will not be described in this tutorial. The work folder also contains all the files that allow the program to be rerun from the break point in case of interruption
Solving proteinase K with the spherical mode in ARCIMBOLDO_SHREDDER
Step by Step tutorial
1. Selection of a starting template
The structure of the proprotein convertase furin from homo sapiens (5JXG), with a 19% of sequence identity to the target proteinase K.
Required Input
We will need:
All required files can be downloaded here.
The description of the configuration file follows:
[CONNECTION]: distribute_computing: local_grid setup_bor_path: /path/to/setup.bor [LOCAL] path_local_phaser: /path/to/local_phaser path_local_shelxe: /path/to/local_shelxe [GENERAL]: working_directory: /path/to/working_directory mtz_path: %(working_directory)s/6v8r.mtz hkl_path: %(working_directory)s/6v8r.hkl [ARCIMBOLDO-SHREDDER] name_job: 6v8r_spheres molecular_weight: 28900 i_label: IMEAN sigi_label: SIGIMEAN formfactors: FORMFACTORS ELECTRON number_of_component: 1 model_file: /absolute/path/to/5jxg.pdb SHRED_METHOD: spherical rmsd_shredder: 0.8 shelxe_line = -m15 -a8 -s0.5 -v0 -t10 -q -o -y1.60 -e1.6 -I15 shelxe_line_last = -m15 -a1 -s0.45 -v0 -t10 -q -o -y1.60 -e1.10 -I15
In the .bor file you need to specify:
In the [ARCIMBOLDO-SHREDDER] section you will need to define the contents of the asymmetric unit, the model pdb to use, and the labels for the mtz file. Other parameters that have defaults but can be changed are the expected rmsd of the models (rmsd_shredder), the definition of their size (sphere_definition), and the settings of model refinement strategies such as gyre and gimble. In this case, we will use default values for all of them. A complete description of all optional and mandatory parameters can be found in the manual, as well as when typing -b option in ARCIMBOLDO_SHREDDER.
Execution
You can run the program from the ccp4i interface, from the XDSGUI or from a terminal, redirecting the output to a log file. This tutorial describes use through a command file, as interfaces are self-explanatory.
1. Interactively
ARCIMBOLDO_SHREDDER 6v8r_spheres.bor
2. In background
nohup ARCIMBOLDO_SHREDDER 6v8r_spheres.bor >& log &
Output and Results
In the directory where you launched ARCIMBOLDO_SHREDDER, you will find a directory called models containing a library of pdbs. Around each Calpha in the template, models are cut in a spatial way, producing a set of non-redundant, overlapping, compact models. In the default mode, they are also annotated in different chains in order to decompose them and perform gyre and gimble refinement, aiming to give more degrees of freedom and obtain a more accurate model.
The directory called ARCIMBOLDO_BORGES contains the output of the ARCIMBOLDO_BORGES run using the library in models. The html output of the library search is found here. The first section echoes all parameters used for the run so that defaults are listed along with values for the parameters set through the .bor file. This allows reproducing the run even if defaults may change in future versions. The next section displays a graph and a table summarizing the rotation clustering step as in ARCIMBOLDO_BORGES. As it can be observed, the rotation cluster identified as 0 has the top figures of merit and the larger number of rotations.
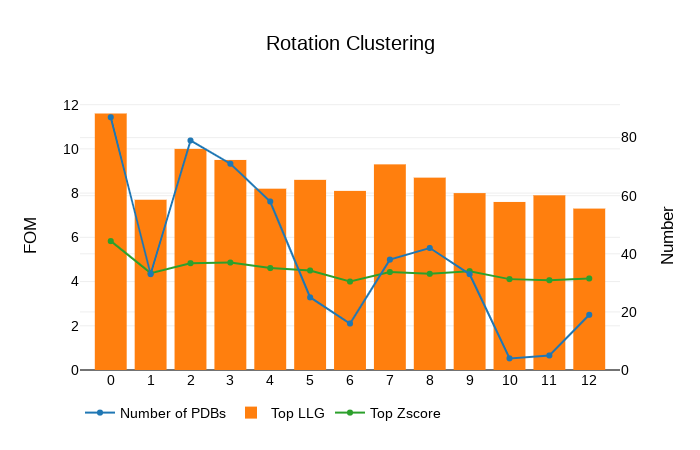
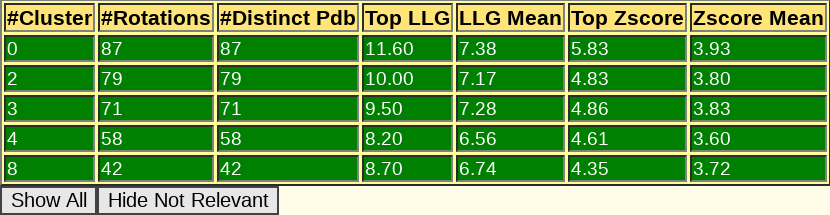
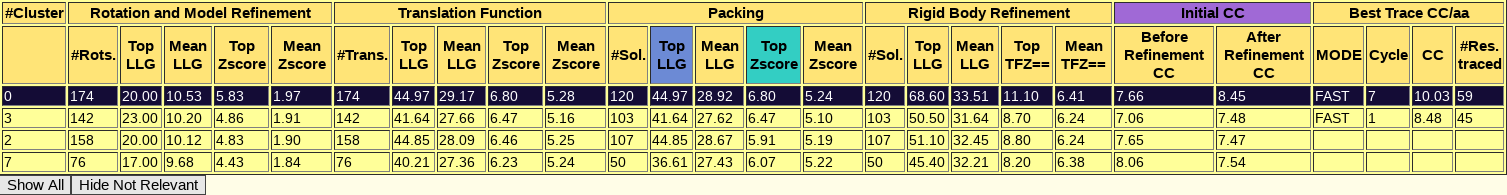
A sortable table follows, summarizing the results for all PHASER and SHELXE steps, including top and average figures of merit for each rotation cluster that has been evaluated. In this table, it is even more clear that rotation cluster 0 is significantly better than the other ones, as it presents the best figures of merit surviving the packing check and in the section of the rigid body refinement, where the gimble refinement has allowed for refinement of parts of the model separated as independent rigid bodies, and the VRMS has also been allowed to refine, the cluster is now clearly better, presenting a top LLG of 68.60, more than 15 points more than any other rotation cluster.
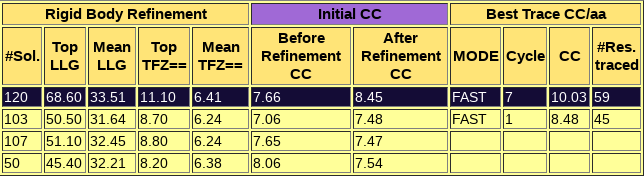
The top solution for which the backtracking is shown, frag255_0_0.pdb is indeed a correct solution, characterised by a wMPE of 73.7º to the true phases of 6V8R. Other 5 solutions from the same rotation cluster have wMPE below 77º. As in the previous case, the SHELXE expansion through density modification and autotracing is not completed successfully in an automatic manner, so the complete building and refinement of the structure starting from this best solution involves a few more steps that will not be described in this tutorial.
References
-
Measuring and using information gained by observing diffraction data.
Read, R. J., Oeffner, R. D. and McCoy, A. J.
Acta Cryst. D76, 238-247 (2020) (doi:10.1107/S2059798320001588)
-
Staraniso.
Tickle, I.J., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., Vonrhein, C. and Bricogne, G.
United Kingdom: Global Phasing Ltd (http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi)
-
Fragment-based determination of Proteinase K structure from MicroED data using ARCIMBOLDO_SHREDDER.
Logan S. Richards, Claudia Millán, Jennifer Miao, Michael W. Martynowycz, Michael R. Sawaya, Tamir Gonen, Rafael J. Borges, Isabel Usón and Jose A. Rodriguez.
Acta Cryst. (2020), submitted