ARCIMBOLDO_SHREDDER tutorial
Aims of the tutorial
This tutorial shows how to create a library of models with ARCIMBOLDO-SHREDDER. SHREDDER evaluates a model template against the rotation function, generates ad-hoc models and uses them within ARCIMBOLDO to solve the structure. This example uses the latest ARCIMBOLDO released on November 2016.
Data tutorial
Experimental details
Test data for our tutorial have been obtained from the protein MltE (PDB ID: 2Y8P). It is a bacterial outer membrane-anchored endolytic peptidoglycan lytic transglycosylase. This structure was solved with data extending to 2.0 Å.
Data are summarized in the following table:

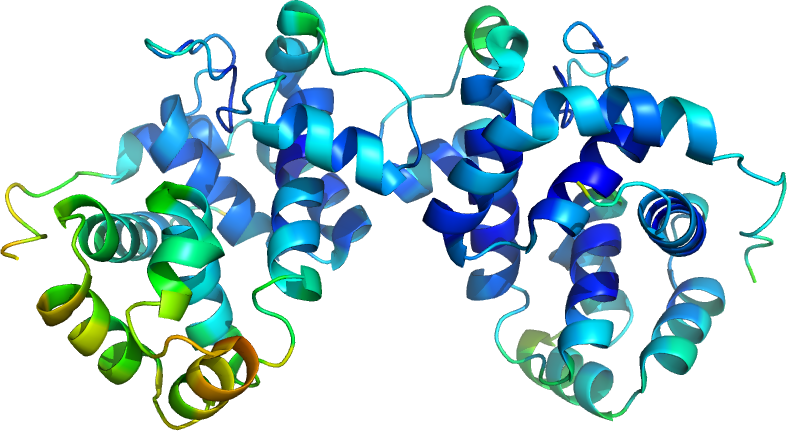
Figure 1. MltE Structure colored after B-value.
Step by Step tutorial
1. Creation of the library from a selected homology model
BLAST search of the MltE sequence against PDB can be used to find a model, 1QTE, with similarity of predicted fold and 35% of identity over a range of 66 amino acids.
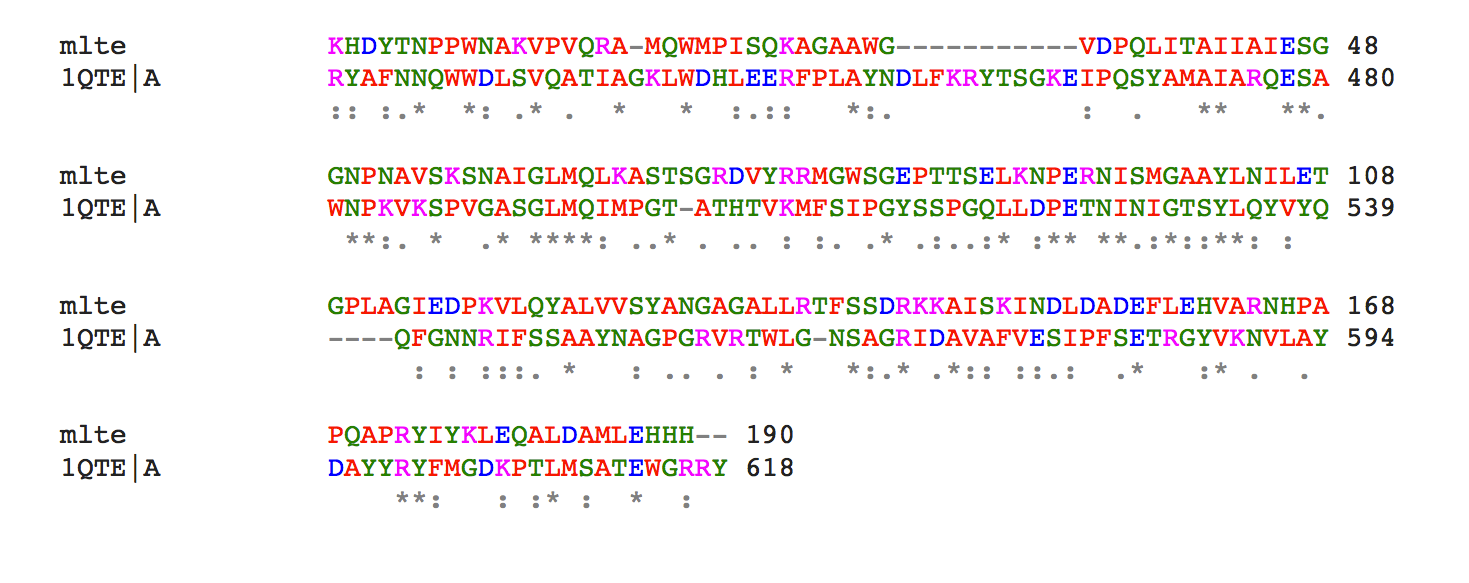
Figure 2. MltE alignment against 1QTE
A classical molecular replacement approach with the 1QTE model trimmed in standard ways fails due to several local structural differences.
Input
We will need:
All required files can be downloaded here.
The description of the configuration file follows:
[CONNECTION]: distribute_computing: local_grid setup_bor_path: /path/to/setup.bor [LOCAL] path_local_phaser: /path/to/local_phaser path_local_shelxe: /path/to/local_shelxe [GENERAL]: working_directory: /path/to/working_directory mtz_path: %(working_directory)s/2y8p.mtz hkl_path: %(working_directory)s/2y8p.hkl [ARCIMBOLDO-SHREDDER] name_job: 2y8p molecular_weight: 20000 i_label: IMEAN sigi_label: SIGIMEAN number_of_component: 2 rmsd_shredder: 1.2 rmsd_arcimboldo: 0.6 model_file: /absolute/path/to/1qte.pdb shelxe_line: -m20 -v0 -a5 -t10 -q -s0.5 -u2000 pack_clashes: 6 fragment_to_search: 2 SHRED_LLG: True SHRED_METHOD: sequential SHRED_RANGE: 4 19 1 omit all
In the .bor file you need to specify:
In the [ARCIMBOLDO-SHREDDER] you will need to define the contents of the asymmetric unit, the model pdb to use, the number of copies to search and the labels for the mtz file. In this case, we also have identity values (as rmsd) to define to PHASER: one to be used in the Shred-LLG evaluation and optimization and one for the ARCIMBOLDO runs launched with the models generated in the first step. Considering that models have been improved and reduced in size with respect to the original template, it is advisable to use a smaller value for the rms deviation for ARCIMBOLDOs than for SHREDDER. We can also set a number of clashes to be allowed in the packing analysis for the ARCIMBOLDO’s runs.
The key part of this section is the one defining the shredding, which in this case will be a sequential shred from 4 to 19 residues with step size is 1. As we input the keyword omit, models will be generated by omitting shreds of 4,5,6....19 residues at every possible starting position.
Execution
You can run the program interactively, getting the output displayed on the screen and inputting the password manually. Alternatively, run in background, redirecting an input file with the passwords you need and passing the output to a log file.
1. Interactively
ARCIMBOLDO_SHREDDER 2y8p.bor
2. In background
nohup ARCIMBOLDO_SHREDDER 2y8p.bor >& log &
Output and Results
In the directory where you launched ARCIMBOLDO_SHREDDER, you have a ./library/ folder containing all models and a set of folders called ARCI_*/, where * refers to the number of the rotation cluster. Inside each folder, there is another set of sub-folders with ARCIMBOLDO runs that are called as the models that have been used. You will find inside of each of them an html file summarising the ARCIMBOLDO run. For our test case, we find that the model percentile75 has been able to solve the structure at the second fragment search. You can check the html output here.
The first section echoes all parameters used for the run, so that defaults are listed along with those set in the .bor file. This allows to reproduce the run even if defaults may change in future versions.The next section displays a sortable table summarizing the results for each step.
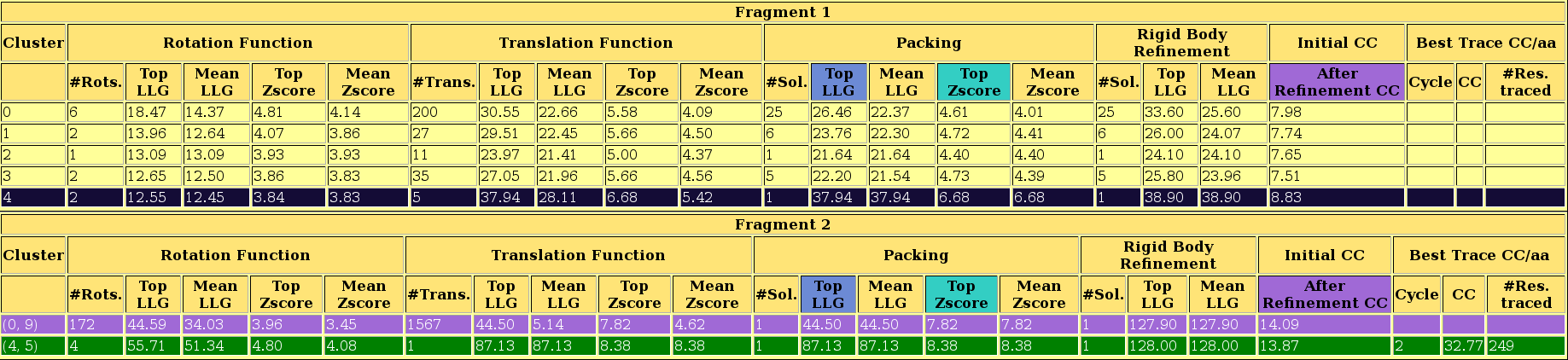
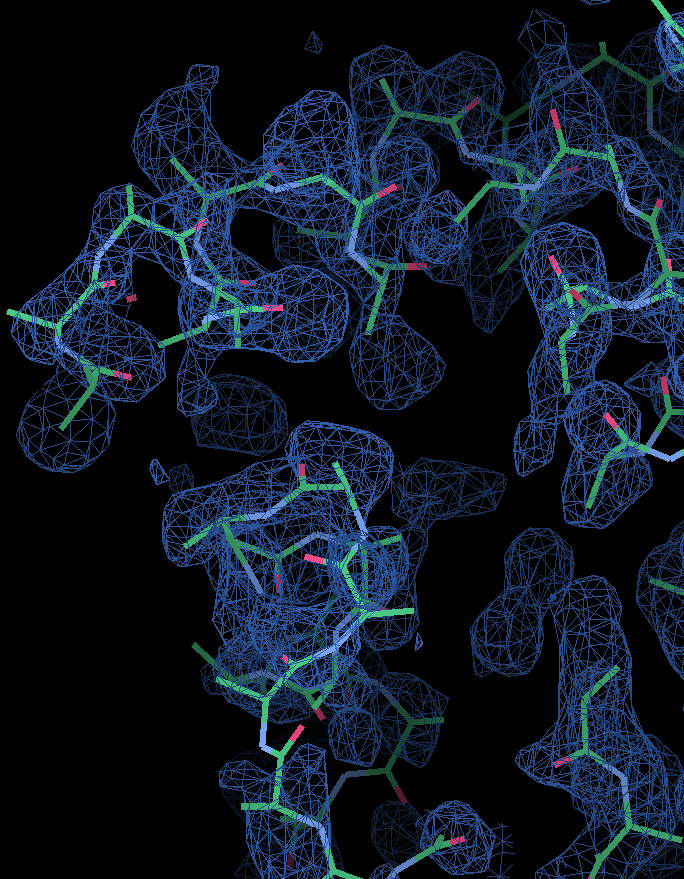
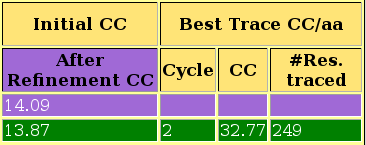
As you can observe in the html file, in this case the structure is solved after location of the second fragment, with a CC of 45.23 for 299 residues traced. The last section of the html contains links to access the best current solution: the pdb of the traced structure and the map in .phs format. The map is interpretable, and the model can be improved to the full trace. The html file also lists the backtracing for the best solution.
The work folder also contains all the files, that allow the program to be rerun from the break point in case of interruption.