ARCIMBOLDO_LITE tutorial
Aims of the tutorial
In this tutorial we will show how to launch ARCIMBOLDO_LITE in order to solve ab initio a 360 aminoacids helical structure at 2.1 Å and analyze the output of the program. This example uses the latest ARCIMBOLDO_LITE released on November 2016.
Data tutorial
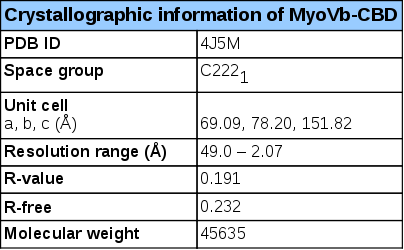
Experimental details – human MyoVb-CBD- structure (4J5M)
The myosin Vb cargo-binding domain (MyoVb-CBD) is the globular tail domain that within the myosin Vb motor protein is responsible for direct interaction with the cargo. MyoV-CBDs are helical domains that display a structural scaffold consisting of two four-helical bundles connected by a long α-helix. The fold is stabilized by a C-terminal extension and a small β-sheet formed by the N- and C-termini of this domain1. The secondary structure prediction (obtained from PSI-PRED server)2 is consistent with this topology (Figure 1).
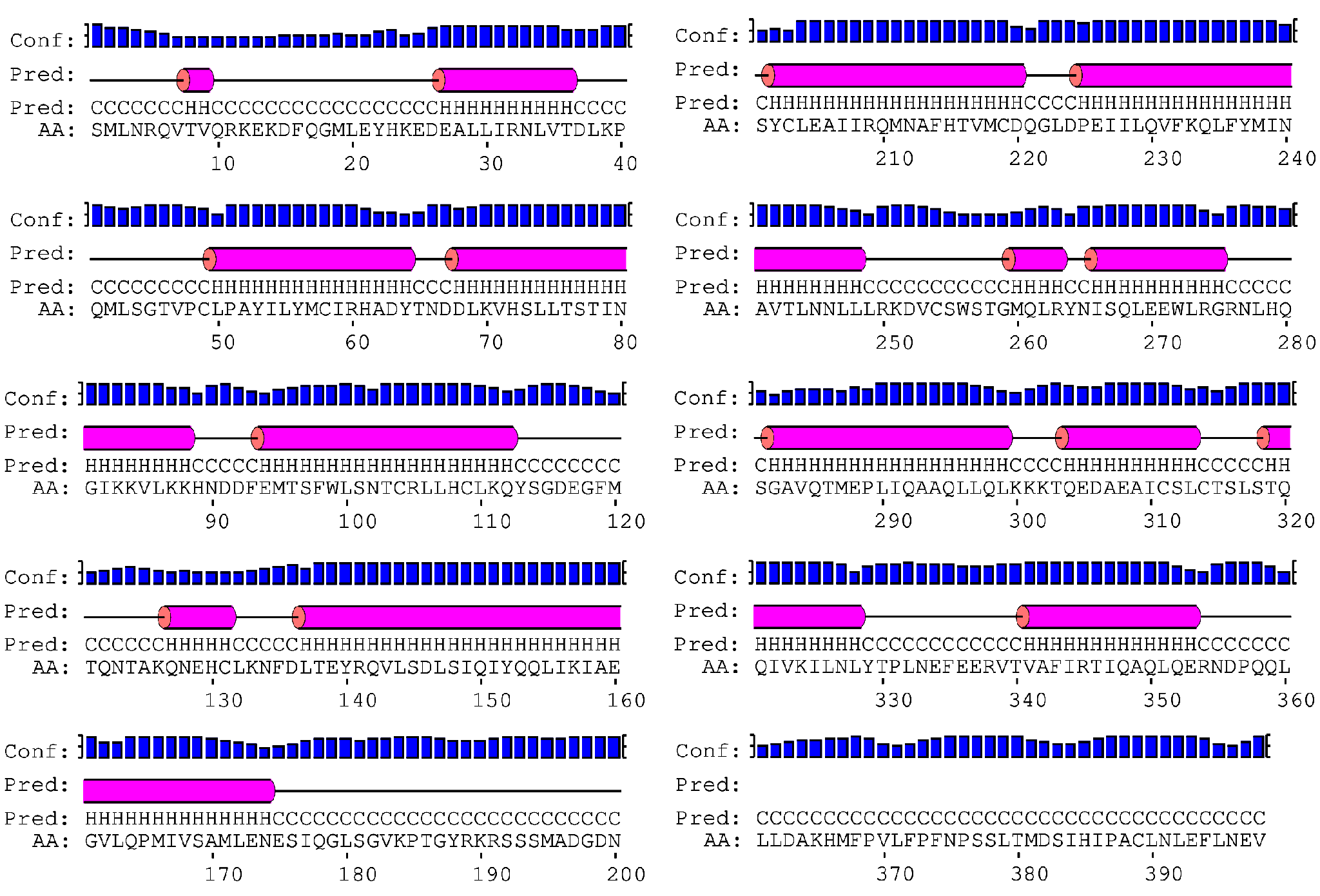
Figure 1. PSI-PRED secondary structure prediction for the MyoVb-CBD sequence. It shows that MyoVb-CBD is mainly composed by α-helices including four 22 residue long helices.
To run ARCIMBOLDO_LITE any source of structural information can be used to define an initial hypothesis, such as secondary or tertiary structure predictions. Information on the folds obtained by biophysical techniques (e.g. circular dichroism) or functional data (presence of some functional conserved motif / domain) can also be exploited to base an initial approach.
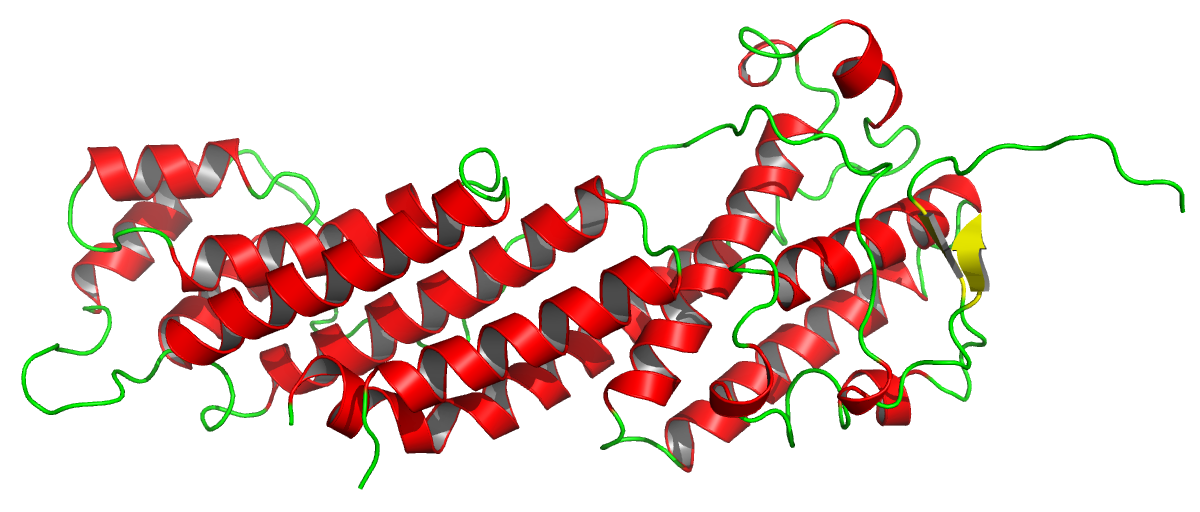
Figure 2. The crystal structure of MyoVb-CBD, 4J5M. The asymmetric unit contains one molecule with 363 residues. The structure is shown highlighting the secondary structure (red, α-helices; beta-strands, yellow; and coils, green). The presence of the several long and straight helices will be the base for the structure solution with ARCIMBOLDO_LITE in this case. This figure was prepared using Pymol3.
Step by Step tutorial
For this tutorial we will need only the reflection file in two formats (.mtz and .hkl). All required files can be downloaded here. The model to search will be an ideal helix that is defined internally, in the program just from its length. For other uses of ARCIMBOLDO_LITE, you may input any model in PDB format. For example, you can provide a specific model, such as a helix extracted from another structure with the side chains. If you only have one of the required reflection file formats, you can use the CCP44 programs MTZ2VARIOUS and F2MTZ to convert an .mtz file to .hkl and an .hkl file to .mtz, respectively. After downloading ARCIMBOLDO_LITE (see instructions here), you are ready to follow this tutorial.
Input
We need 3 files in order to run ARCIMBOLDO_LITE:
Configuration .bor file:
The instructions needed to define a search within ARCIMBOLDO_LITE (such as reflection data, molecular weight and other parameters) should be given in a configuration .bor file. A default .bor file is provided with ARCIMBOLDO_LITE and can be used to run the program. You just need to provide specific values for your case and may leave general parameters at their default values, as shown below.
The .bor file will look like follows:
[CONNECTION]: distribute_computing: multiprocessing working_directory: /path/to/setup.bor [GENERAL]: working_directory: /path/to/working_directory mtz_path: %(working_directory)s/4j5m.mtz hkl_path: %(working_directory)s/4j5m.hkl [ARCIMBOLDO] name_job: 4j5m molecular_weight: 45635 number_of_component: 1 f_label: FOBS sigf_label: SIGFOBS shelxe_line = -m10 -a8 -s0.6 -v0 -u2999 -t10 -q -y2.07 fragment_to_search: 4 helix_length: 22 [LOCAL] path_local_phaser: /path/to/phaser path_local_shelxe: /path/to/shelxe
The [CONNECTION] section contains the information about the type of run and about the general configuration instructions found in the setup.bor file. In this case, we are not going to use a grid but a single machine, in multiprocessing.
Some parameters are particular to the case and you will need to define them, such as local paths.
Parameters for the molecular replacement steps such as definition of the contents of the asymmetric unit must be setup. As we have seen that there are four long helices both in our structure and in the prediction, we will give the variable fragment_to_search a value of four copies of our search model, that it is defined with the parameter helix_length. This means that ARCIMBOLDO_LITE will search two times for an ideal polyalanine helix of 22 residues.
A command line for the SHELXE5 step of the algorithm can be defined. If no shelxe line is set, sensible resolution-dependent default values will be used, but you can change them if required.
Execution
You can run the program interactively, getting the output displayed on the screen and inputting the password manually. The other option is to do it in background, redirecting an input file with the passwords you need and passing the output to a log file.
1. Interactively:
ARCIMBOLDO_LITE 4j5m.bor
2. In background:
nohup ARCIMBOLDO_LITE 4j5m.bor >& logfile.log&
Output and Results
ARCIMBOLDO_LITE will generate several files related to the PHASER6 and SHELXE5 runs. Per each step in the search (rotation, translation, packing, etc), there will be a folder with the output. An overall analysis and a summary of the run can be found in the html output produced. This file is updated during the run and can be opened in an internet browser (Google-Chrome, Firefox, etc.). It summarizes values for the figures of merit in all the steps.The program will save files corresponding to the best scoring solution, whether solved or not, in the working directory:
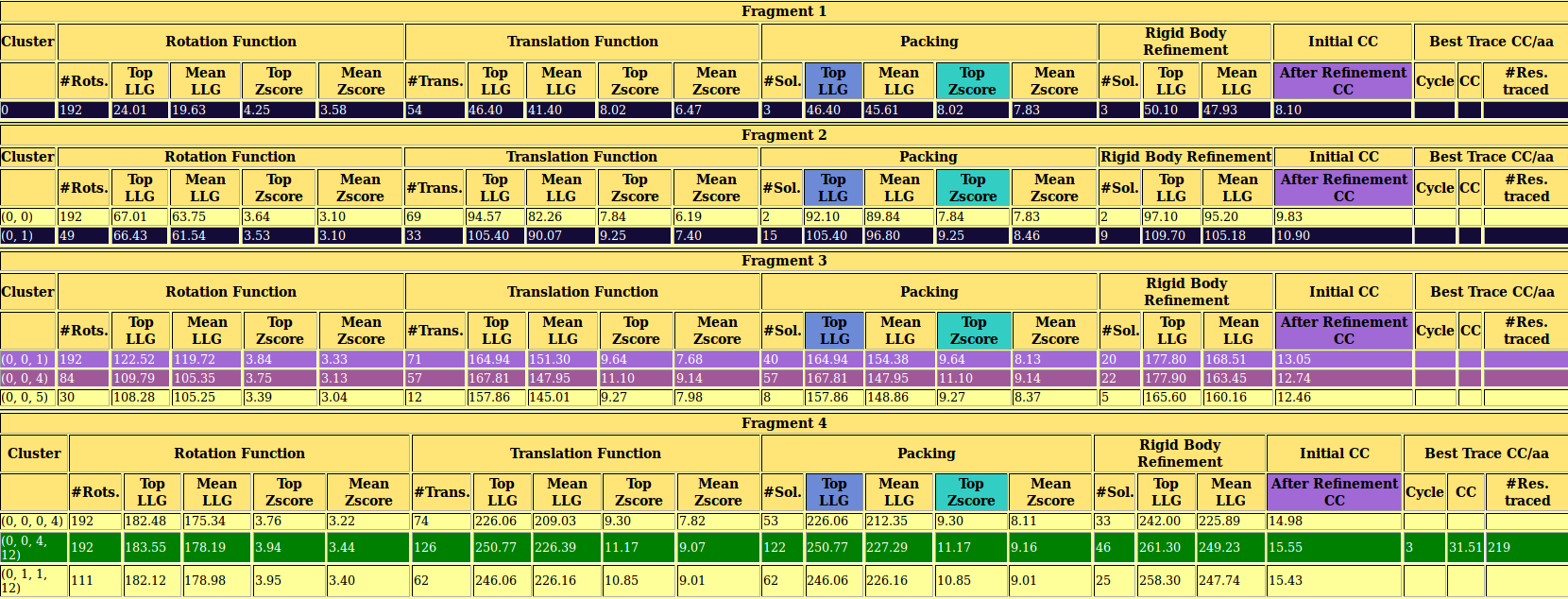
Fig 3. The 4j5m.html. In this file you can find a summary of the run and figures of merit from PHASER6 and SHELXE5, such as RFZ, TFZ, LLG, CC, tracing statistics. This file can be reloaded during the run (reload button of your browser) to check the results interactively. You can open this file here.
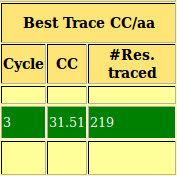
As you can observe in the html file, in this case the structure is already solved after location of the fourth fragment, with a high TFZ and initial CC. By default the program stops after finding a solution with CC over 30%. At the bottom of the file you can find the links to the .pdb of the best solution and its map (.phs). The structure traced and map are shown in Figure 4.
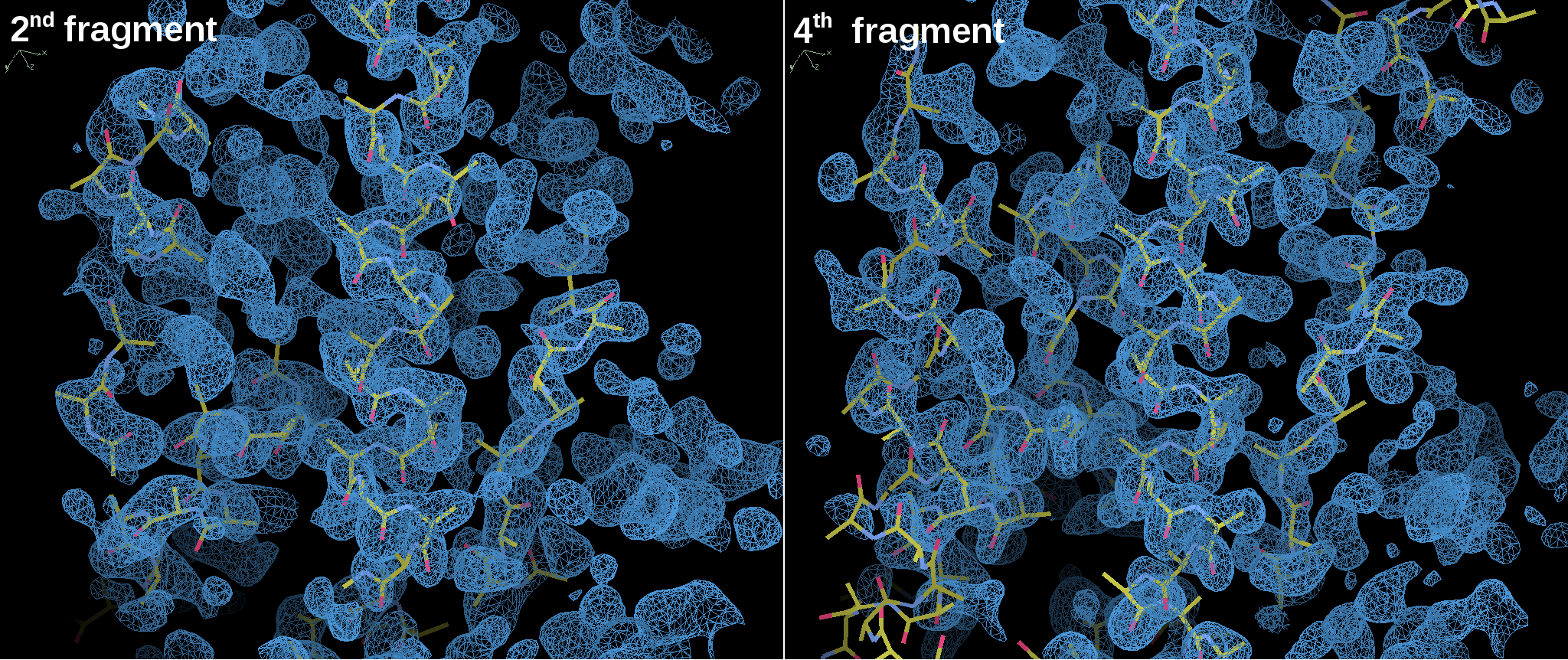
Figure 4. Structure traced and maps for MyoVb-CBD structure solution showed in COOT7.
The main-chain traced by SHELXE5 shows a good agreement with the electron density map and some side-chains can already be identified at this step.
References
-
Crystallographic ab initio protein structure solution below atomic resolution.
Rodríguez, D.D.; Grosse, C.; Himmel, S.; González, C.; Ilarduya, I.M.; Becker, S.; Sheldrick, G.M. and Usón, I.
Nature Methods. 6 (9), 651-653 (2009) (doi: 10.1038/nmeth.1365)
-
Structural insights into functional overlapping and differentiation among myosin V motors.
Nascimento, A.F.Z. et al.
J. Biol. Chem. 288, 34190-34204 (2013) (doi: 10.1074/jbc.M113.507202)
-
Scalable web services for the PSIPRED Protein Analysis Workbench.
Buchan, D.W.A. et al.
Nucleic Acids Res. 41, W349-357 (2013) (doi: 10.1093/nar/gkt381)
-
The PyMOL Molecular Graphics System.
Version 1.5.0.4 Schrödinger, LLC. (https://pymol.org)
-
Overview of the CCP4 suite and current developments.
Winn, M.D. et al.
Acta Cryst. D67, 235-242 (2011) (doi: 10.1107/S0907444910045749)
-
Experimental phasing with SHELXC/D/E: combining chain tracing with density modification.
Sheldrick, G.M.
Acta Cryst. D66, 479-485 (2011) (doi: 10.1107/S0907444909038360)
-
Phaser crystallographic software.
McCoy, A.J. et al.
J. Appl. Crystallogr. D40, 658-674 (2007) (doi: 10.1107/S0021889807021206)
-
Features and development of Coot.
Emsley, P. et al.
Acta Cryst. D66, 486-501 (2010) (doi: 10.1107/S0907444910007493)